


Autor |. Tao Kehao
Herkömmliche Ab-initio-Molekulardynamiksimulationen (AIMD) bieten jedoch natürlich hochpräzise Vorhersagemöglichkeiten, schränken jedoch aufgrund ihres hohen Rechenaufwands und der langen Simulationszeit den Forschungsfortschritt erheblich ein.
Zum Beispiel dauert es normalerweise mehrere Monate, eine 30-Pikosekunden-Simulation eines Materialsystems mit 100 Atomen zu erstellen, was eine große Herausforderung für die Entwicklung neuer Materialien darstellt, die eine schnelle Iteration und Optimierung erfordern.
In diesem Zusammenhang ist ein Modell der künstlichen Intelligenz, das diesen Prozess deutlich beschleunigen kann, von großem Wert.
Angesichts dieser Herausforderungen hat das Labor für künstliche Intelligenz und Mikrostruktur der Shanghai Jiao Tong University (AIMS-Lab) ein revolutionäres Modell für künstliche Intelligenz namens T-AIMD entwickelt.
Dieses Modell verwendet eine transformatorbasierte Netzwerkarchitektur, die nicht nur die Rechenkosten effektiv senkt, sondern auch das Verhalten jedes Ions in jeder Kristallstruktur schnell und genau vorhersagt.
Auf diese Weise beschleunigt das T-AIMD-Modell die herkömmliche AIMD-Simulation um mehr als das Hundertfache und beschleunigt damit den Prozess der Materialleistungsbewertung erheblich.
Darüber hinaus hat das Modell erfolgreich eine große Datenbank gemischter Ionenleiter aufgebaut und die Genauigkeit seiner Vorhersagen in mehreren Batterieexperimenten überprüft.
Diese Methode hat ein breites Anwendungspotenzial nicht nur in den Bereichen Molekulardynamikmodellierung (MD), biopharmazeutische Molekülbindungsziele, Proteinfaltung, thermodynamische Materialprozesse und Berechnung mechanischer Eigenschaften.
Außerdem werden neue Methoden zur Verwendung generativer künstlicher Intelligenzmodelle zur Lösung komplexer Probleme in einem breiteren Spektrum wissenschaftlicher Bereiche bereitgestellt.
Die erfolgreiche Anwendung von T-AIMD zeigt das große Potenzial der Technologie der künstlichen Intelligenz bei der Förderung wissenschaftlicher Forschung und technologischer Innovation und eröffnet neue Wege für die zukünftige Forschung und Entwicklung neuer Materialien sowie die Entwicklung biologischer Designs.
Die Forschung trug den Titel „
Transformer ermöglicht die Entwicklung des Ionentransportverhaltens und die Leitfähigkeitsregulierung für Festelektrolyte“ und wurde am 11. Juni 2024 in der international renommierten Fachzeitschrift „Energy Storage Materials“ veröffentlicht. Der Erstautor des Papiers ist Tao Kehao, ein Doktorand im Labor für künstliche Intelligenz und Mikrostruktur der Shanghai Jiao Tong University, und der korrespondierende Autor ist Professor Li Jinjin, Direktor des Labors.
 Artikellink:
Artikellink: Im Bereich der künstlichen Intelligenz hat sich das Transformer-Modell zum bevorzugten Framework für die Verarbeitung komplexer Sequenzdaten entwickelt .
Dieses Modell eignet sich besonders gut zum Erlernen tiefer Muster und Assoziationen aus großen Datenmengen und wird daher häufig in der Sprachverarbeitung, Bilderkennung und verschiedenen Vorhersageaufgaben eingesetzt.
Dennoch wurde das Potenzial von Transformer in Anwendungen in der Materialwissenschaft, insbesondere in Ab-initio-Molekulardynamiksimulationen (AIMD), noch nicht vollständig ausgeschöpft.
Traditionelle AIMD-Simulationen sind in der Materialwissenschaft sehr wichtig, da sie das dynamische Verhalten von Atomen und Molekülen genau simulieren können. Allerdings beruhen solche Simulationen häufig auf wiederholten Berechnungen und aufwendigen Experimenten, die nicht nur zeitaufwändig, sondern auch kostspielig sind.
Angesichts solcher Herausforderungen ist ein intelligentes Modell, das große Mengen an Sequenzdaten schnell extrahieren und verarbeiten kann, besonders wichtig.
Als Reaktion auf diese Nachfrage nutzt das vom AIMS-Laborteam der Shanghai Jiao Tong University entwickelte T-AIMD-Modell die Transformer-Netzwerkarchitektur, um die Geschwindigkeit und Genauigkeit der AIMD-Simulation deutlich zu verbessern.
Dieses neue Modell kann das Verhalten von Atomen und Molekülen unter verschiedenen Bedingungen schnell und genau analysieren und vorhersagen und gleichzeitig die Rechenkosten erheblich reduzieren.
Im Vergleich zur herkömmlichen AIMD-Methode kann T-AIMD die Simulationsgeschwindigkeit um mehr als das Hundertfache steigern, während gleichzeitig eine hohe Vorhersagegenauigkeit erhalten bleibt und der Materialentwicklungszyklus erheblich verkürzt wird.
Dies stellt nicht nur neue Werkzeuge für die Forschung im Bereich der Materialwissenschaften bereit, sondern demonstriert auch das Anwendungspotenzial von KI bei Hochleistungsrechneraufgaben und eröffnet neue Möglichkeiten für zukünftige wissenschaftliche Erkundungen.
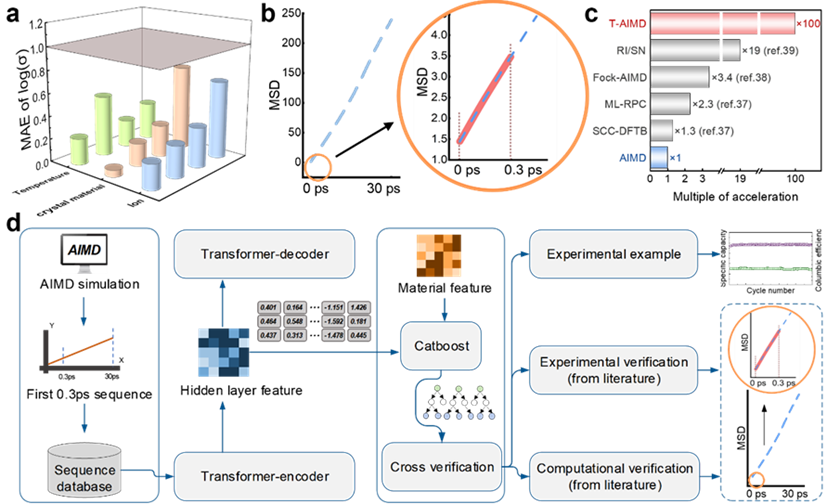 Abbildung: T-AIMD-Vorhersageergebnisse und Workflow-Diagramm. (Quelle: Papier)
Abbildung: T-AIMD-Vorhersageergebnisse und Workflow-Diagramm. (Quelle: Papier)
Nehmen Sie als Beispiel die Lösung des Problems der Vorhersage des Ionentransportverhaltens in Festelektrolyten. Durch das Erlernen der Diffusionssequenz von Ionen im Elektrolyten ist das Modell in der Lage, dessen Verhalten in zukünftigen Zuständen vorherzusagen, was den Prozess der Bewertung von Materialeigenschaften erheblich beschleunigt.
Darüber hinaus enthält das T-AIMD-Modell auch Materialdeskriptoren aus mehreren Quellen, was seine Anwendungsfähigkeit bei der Verarbeitung komplexer Materialsysteme verbessert und es ihm ermöglicht, nicht nur das Verhalten einzelner Ionenarten vorherzusagen, sondern auch Wechselwirkungen in mehreren Ionen zu verarbeiten Systeme Aktion und komplexe Dynamikprobleme.
Diese neue transformatorbasierte Methode bietet eine neue Perspektive und ein neues Werkzeug für die Entwicklung von Festelektrolyten und soll neue Forschungs- und Anwendungsperspektiven im Bereich der Materialwissenschaften eröffnen.
Über die Funktionsweise von T-AIMD
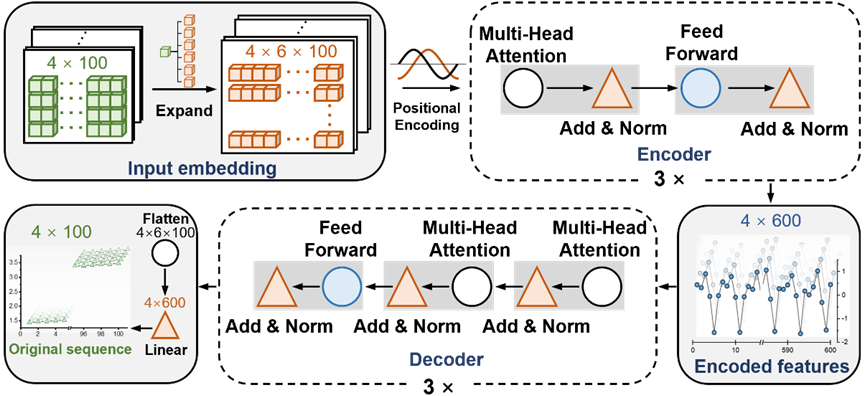
Abbildung: Diagramm der Netzwerkarchitektur von T-AIMD. (Quelle: Paper)
T-AIMD (Transformer-based Ab Initio Molecular Dynamics) ist ein Modell, das Ab-initio-Molekulardynamik (AIMD)-Simulation und Transformer-Deep-Learning-Architektur kombiniert, mit dem Ziel, die Geschwindigkeit des Ionentransports in Festelektrolytmaterialien zu verbessern und Genauigkeit der Vorhersagen der Betriebseigenschaften. Das Funktionsprinzip dieses Modells kann in die folgenden Hauptschritte unterteilt werden:
1. Datenvorbereitung und -vorverarbeitung
T-AIMD sammelt zunächst Ionendiffusionsdaten des Materials, die aus herkömmlichen AIMD-Simulationen stammen. Die durch diese Simulationen generierten Daten umfassen Zeitreihendaten, die die Bewegung von Ionen durch den Elektrolyten aufzeichnen. Diese Sequenzdaten werden vorverarbeitet und in ein Format umgewandelt, das als Eingabe für das maschinelle Lernmodell geeignet ist.
2. Merkmalsextraktion
Mithilfe des Encoder-Teils des Transformer-Modells kann T-AIMD wichtige Merkmale aus Sequenzdaten extrahieren. Dabei erfasst das Modell über einen Selbstaufmerksamkeitsmechanismus Fernabhängigkeiten in der Sequenz, die für das Verständnis komplexer Ionendynamik von entscheidender Bedeutung sind.
3. Sequenzlernen und -vorhersage
Nach der Merkmalsextraktion wird der Decoder-Teil des Transformer-Modells verwendet, um eine Sequenzvorhersage basierend auf den codierten Merkmalen durchzuführen. In diesem Schritt kann das Modell nicht nur das zukünftige Verhalten der Ionen vorhersagen, sondern auch das mögliche Verhalten der Ionen unter verschiedenen Bedingungen, wie beispielsweise unterschiedlichen Temperaturen und Drücken, analysieren. Darüber hinaus kann das Modell anhand dieser erlernten Merkmale wichtige Leistungsindikatoren wie die Ionenleitfähigkeit des Materials vorhersagen.
4. Integration von Materialdeskriptoren aus mehreren Quellen
T-AIMD kombiniert Materialdeskriptoren aus verschiedenen Quellen, wie z. B. Kristallstruktur, Ionenspezies und elektronische Eigenschaften usw., was dem Modell hilft, das Material umfassender zu verstehen und vorherzusagen Eigenschaften. Dieser integrierte Ansatz verbessert die Vielseitigkeit und Anpassungsfähigkeit des Modells in verschiedenen Materialsystemen.
5. Modellüberprüfung und -anwendung
Das entwickelte Modell muss durch Vergleich mit experimentellen Daten und anderen Berechnungsmethoden auf seine Vorhersagegenauigkeit überprüft werden. Nach erfolgreicher Verifizierung kann T-AIMD zum schnellen Screening und Optimieren neuer Zielmaterialien eingesetzt werden, was den Entwicklungszyklus erheblich verkürzt und die Kosten senkt.
Über die robuste Leistung von T-AIMD
Die robuste Leistung des T-AIMD-Modells spiegelt sich hauptsächlich in den folgenden Aspekten wider:
1. Genauigkeit
Das T-AIMD-Modell integriert die Transformer-Architektur, Dadurch wird seine Fähigkeit, komplexe dynamische Verhaltensweisen zu erlernen und vorherzusagen, erheblich verbessert. Im Hinblick auf die AIMD-Simulationsbeschleunigung weist T-AIMD eine höhere Genauigkeit auf als herkömmliche Methoden. Dies ist auf die Anwendung der Deep-Learning-Technologie zurückzuführen, die es dem Modell ermöglicht, das Ionenverhalten auf längeren Zeitskalen in kürzerer Zeit genau vorherzusagen.
2. Recheneffizienz
In Bezug auf die Recheneffizienz ist T-AIMD deutlich besser als die herkömmliche AIMD-Methode. Herkömmliche AIMD-Simulationen nehmen viel Zeit in Anspruch, um die Ionendiffusion zu simulieren, aber T-AIMD reduziert die Abhängigkeit von leistungsstarken Rechenressourcen erheblich, indem es den Berechnungsprozess optimiert und die Simulationszeit von Monaten auf Tage oder Stunden verkürzt.
3. Vielseitigkeit und Flexibilität
T-AIMD kann komplexere Datenstrukturen und größere Datensätze verarbeiten als herkömmliche Modelle für maschinelles Lernen (z. B. Support-Vektor-Maschinen oder Entscheidungsbäume). Das Modell ist in der Lage, sich an viele Arten von Materialien anzupassen und das Verhalten unter verschiedenen Umgebungsbedingungen, wie etwa Temperatur- und Druckänderungen, effektiv vorherzusagen.
4. Modellrobustheit
T-AIMD zeigt eine hohe Robustheit beim Umgang mit Daten mit Rauschen und Unsicherheit. In Vergleichsexperimenten kann T-AIMD ein hohes Maß an Vorhersagegenauigkeit aufrechterhalten, selbst wenn die Daten leicht verzerrt sind, was mit anderen einfachen Modellen für maschinelles Lernen schwer zu erreichen ist.
5. Skalierbarkeit und Anpassungsfähigkeit
Die Architektur des T-AIMD-Modells ermöglicht eine flexible Anpassung und Optimierung zur Anpassung an sich ändernde Forschungsbedürfnisse und neue wissenschaftliche Entdeckungen. Diese Skalierbarkeit ermöglicht es T-AIMD, weiterhin eine Schlüsselrolle in der zukünftigen Forschung zu spielen, wobei sich die Anwendungen über Festkörperelektrolyte hinaus auf die Untersuchung anderer Energiematerialien und komplexer chemischer Systeme erstrecken.
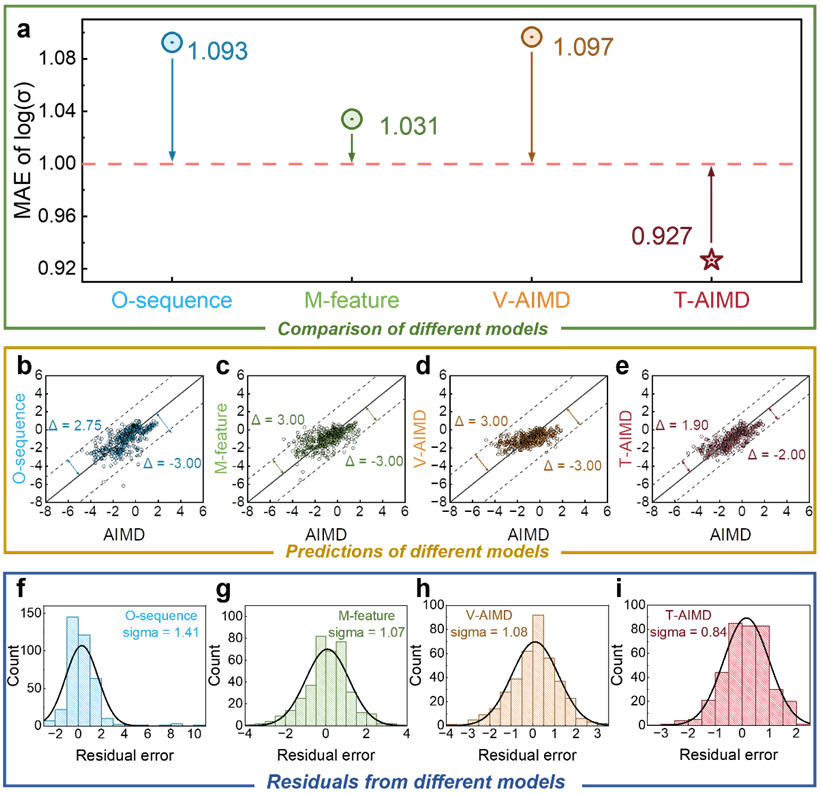
Zusammenfassend lässt sich sagen, dass die Simulationseffizienz der Molekulardynamik basierend auf dem T-AIMD-Framework erheblich beschleunigt werden kann, wodurch die Effizienz um das 1000-fache, 10000-fache oder sogar mehr verbessert wird, was viel Zeit spart Materialherstellung und biologisches Design.
Das T-AIMD-Modell übertrifft herkömmliche AIMD-Simulationen und andere Methoden des maschinellen Lernens in mehreren Schlüsselaspekten, und die im Text aufgeführten Beispiele zeigen sein großes Potenzial und seine Anwendungsaussichten in der Forschung und Entwicklung von Festkörperelektrolyten.
Die Praktikabilität vonT-AIMD geht weit darüber hinaus. Die Leistungsfähigkeit und Flexibilität dieses Modells machen es in vielen Bereichen der Materialwissenschaften weit verbreitet.
In Zukunft soll es zur Vorhersage des Verhaltens von Ionen und Molekülen in anderen Arten von Materialien wie Halbleitern, Metallen und Polymermaterialien eingesetzt werden.
Darüber hinaus ist die Fähigkeit des T-AIMD-Modells nicht auf die Vorhersage des Verhaltens einer einzelnen Ionenart beschränkt. Es kann auch komplexe Wechselwirkungen und Dynamikprobleme in Mehrionensystemen bewältigen, was es bei der Entwicklung neuer Systeme nützlich macht Materialien und die Verbesserung der Leistung vorhandener Materialien. Es hat einen äußerst hohen praktischen Wert.
Das obige ist der detaillierte Inhalt vonDie Recheneffizienz wurde um mehr als das Hundertfache gesteigert und wurde dem Team von Li Jinjin vorgelegt, um ein großes Modell auf Basis von Transformer für Ab-initio-Berechnungen der Molekulardynamik zu entwickeln.. Für weitere Informationen folgen Sie bitte anderen verwandten Artikeln auf der PHP chinesischen Website!
 Wie man unter Linux mit verstümmelten chinesischen Schriftzeichen umgeht
Wie man unter Linux mit verstümmelten chinesischen Schriftzeichen umgeht
 Empfohlene Reihenfolge zum Erlernen von C++ und der C-Sprache
Empfohlene Reihenfolge zum Erlernen von C++ und der C-Sprache
 So lösen Sie verstümmelte Zeichen in PHP
So lösen Sie verstümmelte Zeichen in PHP
 Methode zur Reparatur von Datenbankzweifeln
Methode zur Reparatur von Datenbankzweifeln
 Timeout-Lösung für Serveranfragen
Timeout-Lösung für Serveranfragen
 IIS unerwarteter Fehler 0x8ffe2740 Lösung
IIS unerwarteter Fehler 0x8ffe2740 Lösung
 navigator.useragent
navigator.useragent
 So erhalten Sie ein Token
So erhalten Sie ein Token








![[Web-Frontend] Node.js-Schnellstart](https://img.php.cn/upload/course/000/000/067/662b5d34ba7c0227.png)



