
 Technologie-Peripheriegeräte
KI
In nur wenigen Sekunden können Informationen zur Proteindynamik genau abgeleitet werden. Das KI-Modell RMSF-net der Shandong University, des Beijing Institute of Technology und anderer wurde in der Unterzeitschrift Nature veröffentlicht.
Technologie-Peripheriegeräte
KI
In nur wenigen Sekunden können Informationen zur Proteindynamik genau abgeleitet werden. Das KI-Modell RMSF-net der Shandong University, des Beijing Institute of Technology und anderer wurde in der Unterzeitschrift Nature veröffentlicht.
In nur wenigen Sekunden können Informationen zur Proteindynamik genau abgeleitet werden. Das KI-Modell RMSF-net der Shandong University, des Beijing Institute of Technology und anderer wurde in der Unterzeitschrift Nature veröffentlicht.


Die Dynamik eines Proteins ist entscheidend für das Verständnis seines Mechanismus. Die rechnerische Vorhersage von Informationen zur Proteinkinetik ist jedoch eine Herausforderung.
Hier hat ein Forschungsteam der Shandong University, BioMap, Beijing Institute of Technology, Hubei Medical College, Ningxia Medical University und King Abdullah University of Science and Technology (KAUST) ein neuronales Netzwerkmodell RMSF-net vorgeschlagen, das frühere Methoden übertrifft und liefert die besten Ergebnisse bei umfangreichen Proteindynamikdatensätzen; das Modell kann die Dynamikinformationen eines Proteins in Sekundenschnelle genau ableiten.
Durch effektives Lernen aus der Integration experimenteller Proteinstrukturdaten und Kryo-EM-Daten ist diese Methode in der Lage, interaktive bidirektionale Einschränkungen und Überwachung zwischen Kryo-EM-Bildern und PDB-Modellen genau zu identifizieren, um die Effizienz der Dynamikvorhersage zu maximieren.
RMSF-net ist ein kostenloses Tool, das eine wichtige Rolle bei Studien zur Proteindynamik spielen wird.
Die Studie trug den Titel „Accurate Prediction of Protein Structural Flexibility by Deep Learning Integrating Intricate Atomic Structures and Cryo-EM Density Information“ und wurde am 2. Juli in „Nature Communications“ veröffentlicht.

- https://www.nature.com/articles/s41467-024-49858-x
RMSF-net GitHub-Adresse:
- https://github. com/XintSong/RMSF-net
Proteindynamik
Die Proteindynamik ist entscheidend für das Verständnis ihrer Mechanismen. Die Technologie der Kryo-Elektronenmikroskopie (Kryo-EM) kann die meisten Proteine auflösen, wobei die makromolekulare Struktur durch eine 3D-Dichtekarte dargestellt wird.
Einschränkungen der Kryo-Elektronenmikroskopie
Aufgrund der geringen Auflösung und des Signal-Rausch-Verhältnisses der ursprünglichen 2D-Partikelbilder kann die Kryo-Elektronenmikroskopie-Analyse kleine Konformationsänderungen während des Rekonstruktionsprozesses nicht auflösen.
Anwendung von Deep Learning in der Kryo-Elektronenmikroskopie
Deep-Learning-Methoden werden häufig bei der automatischen Analyse von Bildern der Kryo-Elektronenmikroskopie eingesetzt. Mithilfe hochauflösender Kryo-EM-Karten kann aus den Kryo-EM-Karten ein Proteindatenbankmodell (PDB) erstellt werden.
RMSF-net-Übersicht
RMSF-net ist ein neuronales Netzwerkmodell für Dichtekarten der Kryo-Elektronenmikroskopie. Es nutzt Kryo-EM-Dichte und PDB-Modellinformationen, um in Sekundenschnelle präzise Informationen zur Proteindynamik abzuleiten.
RMSF
RMSF ist eine weit verbreitete Messmethode zur Beurteilung der Flexibilität molekularer Strukturen in molekulardynamischen (MD) Analysen. Sein Hauptzweck besteht darin, den RMSF lokaler Strukturen (Reste, Atome) innerhalb eines Proteins vorherzusagen.
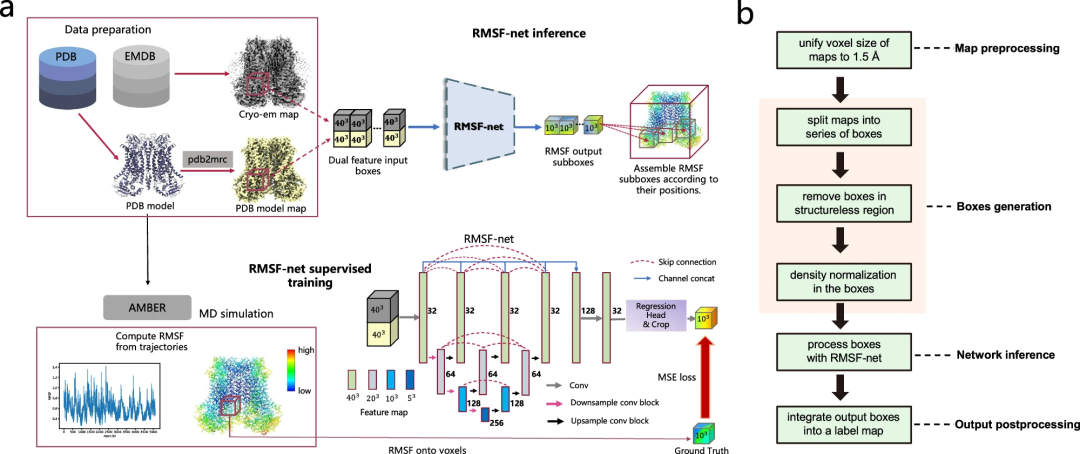
Zusätzlich zu Kryo-EM-Bildern nutzt RMSF-net PDB-Modelle als zusätzliche Eingabe, um RMSF-Vorhersagen zu erstellen, die den MD-Simulationsergebnissen sehr nahe kommen.
RMSF-net ist ein dreidimensionales Faltungs-Neuronales Netzwerk, das zwei miteinander verbundene Module enthält. Das Hauptmodul nutzt die Unet+ (L3)-Architektur, um Funktionen von Eingabedichteboxen zu kodieren und zu dekodieren. Ein anderes Modul nutzt 1x1-Faltungen, um die Kanäle der vom Unet+-Backbone generierten Feature-Maps zurückzuentwickeln. Anschließend wird die Mittenbeschneidung auf die Ausgabe des Regressionsmoduls angewendet, um eine zentrierte RMSF-Unterbox zu erhalten, in der der Voxelwert der RMSF der darin enthaltenen Atome entspricht. Abschließend werden die RMSF-Subboxen mithilfe eines Zusammenführungsalgorithmus räumlich zu einer RMSF-Karte zusammengeführt.
Darüber hinaus erstellten die Forscher auch einen groß angelegten Proteindynamik-Datensatz für das Training und die Validierung von RMSF-net, in dem 335 Kryo-EM-Struktureinträge mit angepassten PDB-Modellen ausgewählt und entsprechende MD-Simulationen durchgeführt wurden. Umfangreiche experimentelle Ergebnisse belegen die Effizienz und Wirksamkeit von RMSF-net.
Tabelle: Leistung verschiedener RMSF-Vorhersagemethoden für den Datensatz. (Quelle: Papier)
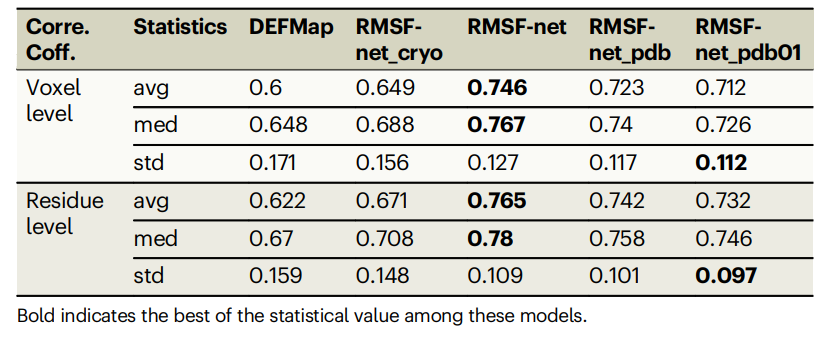
RMSF-net schnitt bei der strengen 5-fachen Kreuzvalidierung gut ab, mit einem Korrelationskoeffizienten von 0,746 ± 0,127 mit MD-Simulationsergebnissen. Der Korrelationskoeffizient von RMSF-net ist im Vergleich zu DEFMap um 15 % und im Vergleich zur Basismethode um 10 % verbessert.
Interpretierbarkeit von Dynamikvorhersagen
Forscher verbesserten die Interpretierbarkeit von RMSF-Netzdynamikvorhersagen durch Vergleichsexperimente. Sie unterteilen den RMSF-Prognoseprozess in zwei Schritte:
- Structural Information Extraction (Occ2RMSF-net)
- Kinematikvorhersage basierend auf extrahierten Strukturinformationen
Studien haben gezeigt, dass die Dynamikvorhersage von Modellen basierend auf Kryo-Elektronenmikroskopie-Spektren (wie DEFMap oder RMSF-net_cryo) hauptsächlich durch erfolgt Interpretation von Proteinen Strukturimplementierung. Dies unterstreicht den Zusammenhang zwischen Proteintopologie und -dynamik im Einklang mit den ersten Prinzipien der Struktur-Funktions-Beziehungen.
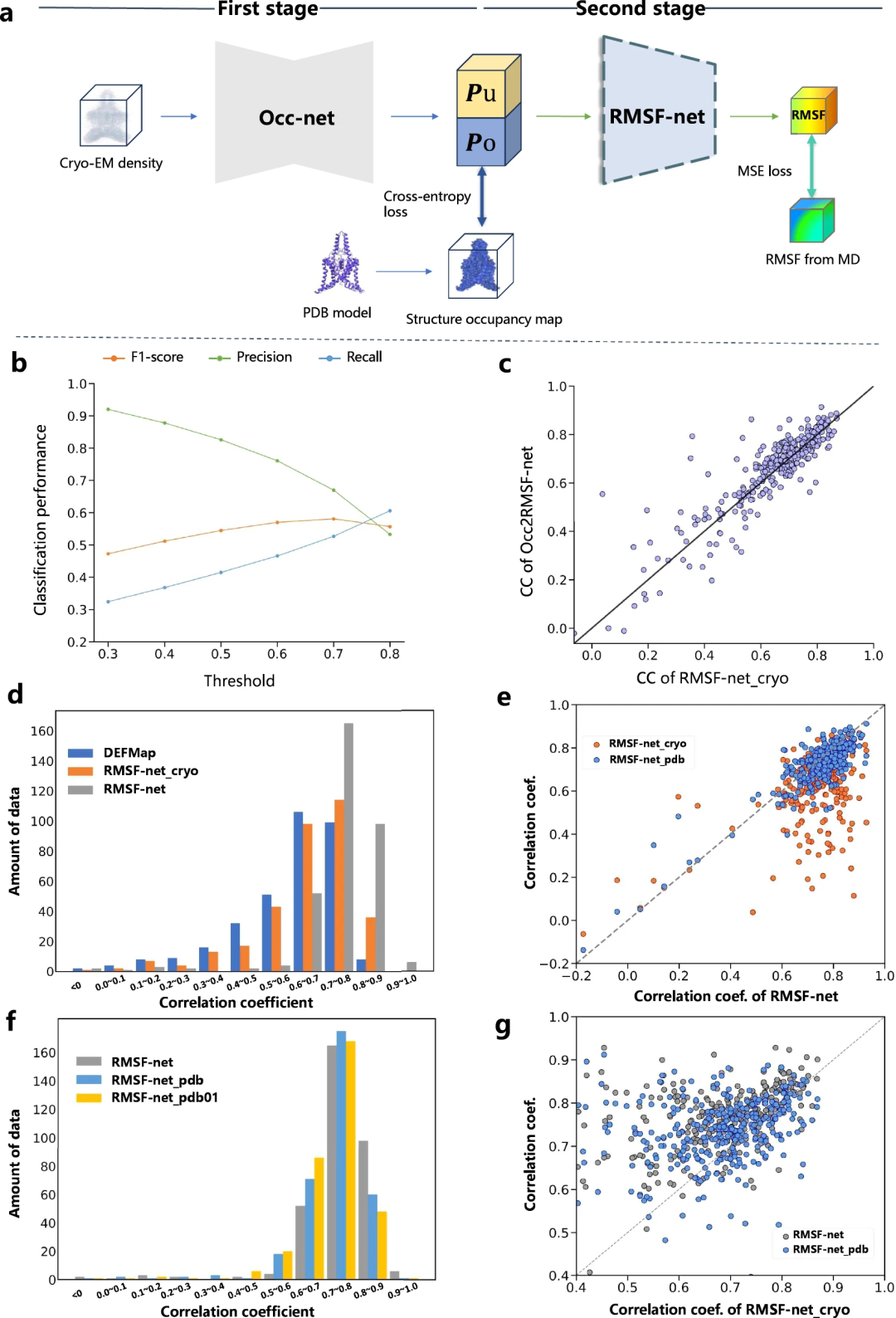
Darüber hinaus wird durch einen umfassenden Vergleich von RMSF-net_cryo, RMSF-net_pdb und der endgültigen Doppelkombination RMSF-net bewiesen, dass: Einerseits die Strukturinformationen aus dem PDB-Modell eine Rolle spielen eine wichtige Rolle in der RMSF-net-Rolle, wo das Tiefenmodell Muster zwischen Strukturtopologie und Flexibilität aus MD-Simulationen lernt, und andererseits wird das Modell durch die kinetischen Informationen, die in der heterogenen Dichteverteilung der Kryo-Elektronenmikroskopie enthalten sind, weiter verbessert Karten. Diese Ergebnisse bestätigen die komplementäre Rolle von Informationen aus Kryo-EM-Karten und PDB-Modellen bei der Vorhersage der Proteindynamik im RMSF-net.
Einschränkungen und zukünftige Richtungen
Es ist unbestreitbar, dass sich RMSF-net hauptsächlich auf die Vorhersage der Flexibilität reiner Proteine und ihrer Komplexe in Lösung beschränkt. Diese Methode kann in bestimmten lokalisierten Regionen Ungenauigkeiten hinsichtlich der dynamischen Eigenschaften des Proteins aufweisen, wenn es an kleine Molekülliganden oder in einer Membranumgebung gebunden ist.
Die hervorragende Leistung von RMSF-net zeigt die Machbarkeit weiterer Forschung in dieser Richtung. Diese Forschung wurde nicht auf Nukleinsäuren und Protein-Nukleinsäure-Komplexe ausgeweitet. Eine umfassende Charakterisierung aller Aspekte der makromolekularen Dynamik, einschließlich Multikonformationsvorhersage und Übergangsanalyse, erfordert in Zukunft weitere umfangreiche und tiefgreifende Forschung.
Dennoch hat RMSF-net als Werkzeug zur Vorhersage der Proteindynamik aufgrund seiner überlegenen Leistung und ultraschnellen Verarbeitungsgeschwindigkeit immer noch große Anwendungsaussichten in der Proteinstruktur- und -dynamikforschung.
Hinweis: Das Cover stammt aus dem Internet
Das obige ist der detaillierte Inhalt vonIn nur wenigen Sekunden können Informationen zur Proteindynamik genau abgeleitet werden. Das KI-Modell RMSF-net der Shandong University, des Beijing Institute of Technology und anderer wurde in der Unterzeitschrift Nature veröffentlicht.. Für weitere Informationen folgen Sie bitte anderen verwandten Artikeln auf der PHP chinesischen Website!

Heiße KI -Werkzeuge

Undresser.AI Undress
KI-gestützte App zum Erstellen realistischer Aktfotos

AI Clothes Remover
Online-KI-Tool zum Entfernen von Kleidung aus Fotos.

Undress AI Tool
Ausziehbilder kostenlos

Clothoff.io
KI-Kleiderentferner

Video Face Swap
Tauschen Sie Gesichter in jedem Video mühelos mit unserem völlig kostenlosen KI-Gesichtstausch-Tool aus!

Heißer Artikel
Heiße Werkzeuge

Notepad++7.3.1
Einfach zu bedienender und kostenloser Code-Editor

SublimeText3 chinesische Version
Chinesische Version, sehr einfach zu bedienen

Senden Sie Studio 13.0.1
Leistungsstarke integrierte PHP-Entwicklungsumgebung

Dreamweaver CS6
Visuelle Webentwicklungstools

SublimeText3 Mac-Version
Codebearbeitungssoftware auf Gottesniveau (SublimeText3)
Heiße Themen



 „Defect Spectrum' durchbricht die Grenzen der herkömmlichen Fehlererkennung und erreicht erstmals eine hochpräzise und umfassende semantische Fehlererkennung in der Industrie.
Jul 26, 2024 pm 05:38 PM
„Defect Spectrum' durchbricht die Grenzen der herkömmlichen Fehlererkennung und erreicht erstmals eine hochpräzise und umfassende semantische Fehlererkennung in der Industrie.
Jul 26, 2024 pm 05:38 PM
In der modernen Fertigung ist die genaue Fehlererkennung nicht nur der Schlüssel zur Sicherstellung der Produktqualität, sondern auch der Kern für die Verbesserung der Produktionseffizienz. Allerdings mangelt es vorhandenen Datensätzen zur Fehlererkennung häufig an der Genauigkeit und dem semantischen Reichtum, die für praktische Anwendungen erforderlich sind, was dazu führt, dass Modelle bestimmte Fehlerkategorien oder -orte nicht identifizieren können. Um dieses Problem zu lösen, hat ein Spitzenforschungsteam bestehend aus der Hong Kong University of Science and Technology Guangzhou und Simou Technology innovativ den „DefectSpectrum“-Datensatz entwickelt, der eine detaillierte und semantisch reichhaltige groß angelegte Annotation von Industriedefekten ermöglicht. Wie in Tabelle 1 gezeigt, bietet der Datensatz „DefectSpectrum“ im Vergleich zu anderen Industriedatensätzen die meisten Fehleranmerkungen (5438 Fehlerproben) und die detaillierteste Fehlerklassifizierung (125 Fehlerkategorien).
 Das NVIDIA-Dialogmodell ChatQA wurde auf Version 2.0 weiterentwickelt, wobei die angegebene Kontextlänge 128 KB beträgt
Jul 26, 2024 am 08:40 AM
Das NVIDIA-Dialogmodell ChatQA wurde auf Version 2.0 weiterentwickelt, wobei die angegebene Kontextlänge 128 KB beträgt
Jul 26, 2024 am 08:40 AM
Die offene LLM-Community ist eine Ära, in der hundert Blumen blühen und konkurrieren. Sie können Llama-3-70B-Instruct, QWen2-72B-Instruct, Nemotron-4-340B-Instruct, Mixtral-8x22BInstruct-v0.1 und viele andere sehen hervorragende Darsteller. Allerdings weisen offene Modelle im Vergleich zu den proprietären Großmodellen GPT-4-Turbo in vielen Bereichen noch erhebliche Lücken auf. Zusätzlich zu allgemeinen Modellen wurden einige offene Modelle entwickelt, die sich auf Schlüsselbereiche spezialisieren, wie etwa DeepSeek-Coder-V2 für Programmierung und Mathematik und InternVL für visuelle Sprachaufgaben.
 Training mit Millionen von Kristalldaten zur Lösung kristallographischer Phasenprobleme, die Deep-Learning-Methode PhAI wird in Science veröffentlicht
Aug 08, 2024 pm 09:22 PM
Training mit Millionen von Kristalldaten zur Lösung kristallographischer Phasenprobleme, die Deep-Learning-Methode PhAI wird in Science veröffentlicht
Aug 08, 2024 pm 09:22 PM
Herausgeber |KX Bis heute sind die durch die Kristallographie ermittelten Strukturdetails und Präzision, von einfachen Metallen bis hin zu großen Membranproteinen, mit keiner anderen Methode zu erreichen. Die größte Herausforderung, das sogenannte Phasenproblem, bleibt jedoch die Gewinnung von Phaseninformationen aus experimentell bestimmten Amplituden. Forscher der Universität Kopenhagen in Dänemark haben eine Deep-Learning-Methode namens PhAI entwickelt, um Kristallphasenprobleme zu lösen. Ein Deep-Learning-Neuronales Netzwerk, das mithilfe von Millionen künstlicher Kristallstrukturen und den entsprechenden synthetischen Beugungsdaten trainiert wird, kann genaue Elektronendichtekarten erstellen. Die Studie zeigt, dass diese Deep-Learning-basierte Ab-initio-Strukturlösungsmethode das Phasenproblem mit einer Auflösung von nur 2 Angström lösen kann, was nur 10 bis 20 % der bei atomarer Auflösung verfügbaren Daten im Vergleich zur herkömmlichen Ab-initio-Berechnung entspricht
 Google AI gewann die Silbermedaille der IMO Mathematical Olympiad, das mathematische Argumentationsmodell AlphaProof wurde eingeführt und Reinforcement Learning ist zurück
Jul 26, 2024 pm 02:40 PM
Google AI gewann die Silbermedaille der IMO Mathematical Olympiad, das mathematische Argumentationsmodell AlphaProof wurde eingeführt und Reinforcement Learning ist zurück
Jul 26, 2024 pm 02:40 PM
Für KI ist die Mathematikolympiade kein Problem mehr. Am Donnerstag hat die künstliche Intelligenz von Google DeepMind eine Meisterleistung vollbracht: Sie nutzte KI, um meiner Meinung nach die eigentliche Frage der diesjährigen Internationalen Mathematikolympiade zu lösen, und war nur einen Schritt davon entfernt, die Goldmedaille zu gewinnen. Der IMO-Wettbewerb, der gerade letzte Woche zu Ende ging, hatte sechs Fragen zu Algebra, Kombinatorik, Geometrie und Zahlentheorie. Das von Google vorgeschlagene hybride KI-System beantwortete vier Fragen richtig und erzielte 28 Punkte und erreichte damit die Silbermedaillenstufe. Anfang dieses Monats hatte der UCLA-Professor Terence Tao gerade die KI-Mathematische Olympiade (AIMO Progress Award) mit einem Millionenpreis gefördert. Unerwarteterweise hatte sich das Niveau der KI-Problemlösung vor Juli auf dieses Niveau verbessert. Beantworten Sie die Fragen meiner Meinung nach gleichzeitig. Am schwierigsten ist es meiner Meinung nach, da sie die längste Geschichte, den größten Umfang und die negativsten Fragen haben
 PRO |. Warum verdienen große Modelle, die auf MoE basieren, mehr Aufmerksamkeit?
Aug 07, 2024 pm 07:08 PM
PRO |. Warum verdienen große Modelle, die auf MoE basieren, mehr Aufmerksamkeit?
Aug 07, 2024 pm 07:08 PM
Im Jahr 2023 entwickeln sich fast alle Bereiche der KI in beispielloser Geschwindigkeit weiter. Gleichzeitig verschiebt die KI ständig die technologischen Grenzen wichtiger Bereiche wie der verkörperten Intelligenz und des autonomen Fahrens. Wird der Status von Transformer als Mainstream-Architektur großer KI-Modelle durch den multimodalen Trend erschüttert? Warum ist die Erforschung großer Modelle auf Basis der MoE-Architektur (Mixture of Experts) zu einem neuen Trend in der Branche geworden? Können Large Vision Models (LVM) ein neuer Durchbruch im allgemeinen Sehvermögen sein? ...Aus dem PRO-Mitglieder-Newsletter 2023 dieser Website, der in den letzten sechs Monaten veröffentlicht wurde, haben wir 10 spezielle Interpretationen ausgewählt, die eine detaillierte Analyse der technologischen Trends und industriellen Veränderungen in den oben genannten Bereichen bieten, um Ihnen dabei zu helfen, Ihre Ziele in der Zukunft zu erreichen Jahr vorbereitet sein. Diese Interpretation stammt aus Week50 2023
 Um ein neues wissenschaftliches und komplexes Frage-Antwort-Benchmark- und Bewertungssystem für große Modelle bereitzustellen, haben UNSW, Argonne, die University of Chicago und andere Institutionen gemeinsam das SciQAG-Framework eingeführt
Jul 25, 2024 am 06:42 AM
Um ein neues wissenschaftliches und komplexes Frage-Antwort-Benchmark- und Bewertungssystem für große Modelle bereitzustellen, haben UNSW, Argonne, die University of Chicago und andere Institutionen gemeinsam das SciQAG-Framework eingeführt
Jul 25, 2024 am 06:42 AM
Herausgeber | Der Frage-Antwort-Datensatz (QA) von ScienceAI spielt eine entscheidende Rolle bei der Förderung der Forschung zur Verarbeitung natürlicher Sprache (NLP). Hochwertige QS-Datensätze können nicht nur zur Feinabstimmung von Modellen verwendet werden, sondern auch effektiv die Fähigkeiten großer Sprachmodelle (LLMs) bewerten, insbesondere die Fähigkeit, wissenschaftliche Erkenntnisse zu verstehen und zu begründen. Obwohl es derzeit viele wissenschaftliche QS-Datensätze aus den Bereichen Medizin, Chemie, Biologie und anderen Bereichen gibt, weisen diese Datensätze immer noch einige Mängel auf. Erstens ist das Datenformular relativ einfach, die meisten davon sind Multiple-Choice-Fragen. Sie sind leicht auszuwerten, schränken jedoch den Antwortauswahlbereich des Modells ein und können die Fähigkeit des Modells zur Beantwortung wissenschaftlicher Fragen nicht vollständig testen. Im Gegensatz dazu offene Fragen und Antworten
 Die Genauigkeitsrate erreicht 60,8 %. Das auf Transformer basierende Modell zur Vorhersage der chemischen Retrosynthese wurde in der Unterzeitschrift „Nature' veröffentlicht
Aug 06, 2024 pm 07:34 PM
Die Genauigkeitsrate erreicht 60,8 %. Das auf Transformer basierende Modell zur Vorhersage der chemischen Retrosynthese wurde in der Unterzeitschrift „Nature' veröffentlicht
Aug 06, 2024 pm 07:34 PM
Herausgeber | KX-Retrosynthese ist eine entscheidende Aufgabe in der Arzneimittelforschung und organischen Synthese, und KI wird zunehmend eingesetzt, um den Prozess zu beschleunigen. Bestehende KI-Methoden weisen eine unbefriedigende Leistung und eine begrenzte Vielfalt auf. In der Praxis verursachen chemische Reaktionen häufig lokale molekulare Veränderungen mit erheblichen Überschneidungen zwischen Reaktanten und Produkten. Davon inspiriert schlug das Team von Hou Tingjun an der Zhejiang-Universität vor, die einstufige retrosynthetische Vorhersage als eine Aufgabe zur Bearbeitung molekularer Ketten neu zu definieren und dabei die Zielmolekülkette iterativ zu verfeinern, um Vorläuferverbindungen zu erzeugen. Außerdem wird ein bearbeitungsbasiertes retrosynthetisches Modell EditRetro vorgeschlagen, mit dem qualitativ hochwertige und vielfältige Vorhersagen erzielt werden können. Umfangreiche Experimente zeigen, dass das Modell beim Standard-Benchmark-Datensatz USPTO-50 K eine hervorragende Leistung mit einer Top-1-Genauigkeit von 60,8 % erzielt.
 Der Standpunkt der Natur: Die Erprobung künstlicher Intelligenz in der Medizin ist im Chaos. Was ist zu tun?
Aug 22, 2024 pm 04:37 PM
Der Standpunkt der Natur: Die Erprobung künstlicher Intelligenz in der Medizin ist im Chaos. Was ist zu tun?
Aug 22, 2024 pm 04:37 PM
Herausgeber | ScienceAI Basierend auf begrenzten klinischen Daten wurden Hunderte medizinischer Algorithmen genehmigt. Wissenschaftler diskutieren darüber, wer die Werkzeuge testen soll und wie dies am besten geschieht. Devin Singh wurde Zeuge, wie ein pädiatrischer Patient in der Notaufnahme einen Herzstillstand erlitt, während er lange auf eine Behandlung wartete, was ihn dazu veranlasste, den Einsatz von KI zu erforschen, um Wartezeiten zu verkürzen. Mithilfe von Triage-Daten aus den Notaufnahmen von SickKids erstellten Singh und Kollegen eine Reihe von KI-Modellen, um mögliche Diagnosen zu stellen und Tests zu empfehlen. Eine Studie zeigte, dass diese Modelle die Zahl der Arztbesuche um 22,3 % verkürzen können und die Verarbeitung der Ergebnisse pro Patient, der einen medizinischen Test benötigt, um fast drei Stunden beschleunigt. Der Erfolg von Algorithmen der künstlichen Intelligenz in der Forschung bestätigt dies jedoch nur
