
 Technologie-Peripheriegeräte
KI
„AlphaFold 3 macht einen wichtigen Schritt in Richtung Entschlüsselung molekularen Verhaltens und biologischer Datenverarbeitung', kommentierte ein „Nature'-Unterjournal
Technologie-Peripheriegeräte
KI
„AlphaFold 3 macht einen wichtigen Schritt in Richtung Entschlüsselung molekularen Verhaltens und biologischer Datenverarbeitung', kommentierte ein „Nature'-Unterjournal
„AlphaFold 3 macht einen wichtigen Schritt in Richtung Entschlüsselung molekularen Verhaltens und biologischer Datenverarbeitung', kommentierte ein „Nature'-Unterjournal


Wenn wir vollständig verstehen würden, wie Moleküle miteinander interagieren, gäbe es nichts mehr über Biologie zu lernen, denn jedes biologische Phänomen, einschließlich unserer Wahrnehmung der Welt, hat letztendlich seinen Ursprung im Verhalten und den Interaktionen von Zellen Biomoleküle im Inneren.
Das kürzlich eingeführte AlphaFold 3 kann die 3D-Struktur biomolekularer Komplexe direkt aus den Sequenzen von Proteinen, Nukleinsäuren und ihren Liganden vorhersagen. Dies stellt einen bedeutenden Fortschritt in unserer langfristigen Erforschung der Interaktion von Biomolekülen dar.
AlphaFold 3 stellt einen Durchbruch bei der Vorhersage der dreidimensionalen Struktur eines Komplexes direkt aus seiner Sequenz dar und liefert Einblicke in biomolekulare Wechselwirkungen.

Eine eindimensionale (1D) Sequenz eines Biomoleküls (z. B. eines Proteins oder einer Nukleinsäure), die eine zelluläre Funktion angibt, ähnlich einem Code, der ein Programm angibt. Diese Sequenz stellt Code in einer Programmiersprache dar und wird durch einen Faltprozess zu Code in Maschinensprache „kompiliert“, wodurch eine einzigartige 3D-Struktur entsteht.
- Ausführung des Programms
Das Programm wird durch die Interaktion zwischen den gefalteten Biomolekülen und anderen Molekülen innerhalb der Zelle ausgeführt.
- Wechselwirkungen von Biomolekülen
Aufgrund ihrer einzigartigen dreidimensionalen Struktur interagieren Biomoleküle nur mit einer kleinen Anzahl von Molekülen innerhalb der Zelle (z. B. DNA-Stellen) und diese Wechselwirkungen lösen eine Reihe sorgfältig geplanter Prozesse aus chemische und strukturelle Transformationen, die zusammen biochemische Programme definieren (z. B. Transkription). Die Produkte biochemischer Prozesse, wie zum Beispiel RNA, stellen den Output des ausführenden Programms dar.
- Kodierung biologischer Sequenzen
In der Biologie kodiert also eine eindimensionale Folge von Biomolekülen das Programm und die Mittel zum Kompilieren und Ausführen des Programms; Die Vorhersage der dreidimensionalen Strukturen, die von Biomolekülkomplexen auf der Grundlage ihrer eindimensionalen Sequenzen gebildet werden, ist ein entscheidender Schritt zum Verständnis der Funktionsweise biologischer Programme, mit tiefgreifenden Auswirkungen auf unsere Fähigkeit, biologische Systeme zu verstehen, rational zu manipulieren und zu entwerfen.

1. AlphaFold 2
- Veröffentlicht im Jahr 2020, ein revolutionärer Algorithmus zur Vorhersage der Proteinstruktur
- Ausgezeichnete mittlere Genauigkeit im Vergleich zu anderen Methoden
- liefert vorhergesagte Strukturen von 200 Millionen bekannten Proteinen Multi-Track-Netzwerke zur Erreichung von hoher Genauigkeit
3
- Veröffentlicht im Mai 2023
- Gehen Sie über professionelle Tools hinaus, um die 3D-Struktur von Proteinkomplexen vorherzusagen
- Erhebliche Verbesserung der Vorhersagegenauigkeit von Protein-Ligand- und Protein-Nukleinsäure-Komplexen
- Prognostizieren Sie Strukturen, die mehrere kovalente Modifikationen enthalten
5. Technologie-Update
- Ersetzen Sie das Strukturmodul durch das Diffusionsmodul.
- Prognostizieren Sie direkt die kartesischen Koordinaten einzelner Atome 3's Diffusionsmodul. (Quelle: Papier)
Als vereinfachte Illustration von AlphaFold 3:
- Stellen Sie sich vor, Sie nehmen die dreidimensionalen Koordinaten jedes Atoms in einem typischen Biomolekülkomplex.
- Fügen Sie iterativ immer mehr Gaußsches Rauschen hinzu, bis wir eine zufällig verteilte Atomwolke im Raum erhalten (Vorwärtsdiffusion).
- Diffusionsmodelle nutzen mehrschichtige neuronale Netze, um zu lernen, diesen Prozess umzukehren (Rückdiffusion).
- Auf diese Weise lernt das Diffusionsmodul in AlphaFold 3:
Die Koordinaten jedes Atoms in einem bestimmten Komplex vorherzusagen, ohne dass ein vordefiniertes Restgerüst erforderlich ist. Umfassenderer chemischer Raum, einschließlich Nukleinsäuren, Ionen, Liganden und chemische Modifikationen.
- Weitere Verbesserungen:
-
Evoformer wurde durch Pairformer ersetzt, eine neuere Transformer-Architektur.
Weniger Schwerpunkt auf MSA-Handhabung. Aktualisieren Sie Metriken, um sie an Änderungen in der Netzwerkarchitektur anzupassen.

Fortschritte und Einschränkungen:
- Fortschritt: Verbesserte Vorhersagegenauigkeit, geringere Abhängigkeit vom Sequenz-Alignment und stärkere Betonung von Restwechselwirkungen.
- Einschränkungen: Manchmal kann die Chiralität von Molekülen nicht korrekt simuliert werden, die Struktur großer Protein-Nukleinsäure-Komplexe kann nicht vorhergesagt werden und das generierte Modell kann „Halluzinationen“ aufweisen.
RNA-Vorhersage:
- Die Vorhersagegenauigkeit von AlphaFold 3 für RNA-Ziele ist höher als bei anderen Methoden, aber nicht so genau wie die von Top-Experten.
AlphaFold Server:
- bietet eine benutzerfreundliche Oberfläche, aber der Quellcode und die ausführbaren Dateien sind nicht öffentlich.
- Pseudocode ersetzt Quellcode, was zu Kontroversen führt und die weitere Entwicklung behindert.
1. Wenn man die durch AlphaFold 3 erzielten Durchbrüche in der Strukturvorhersage betrachtet, ist es wichtig, sich daran zu erinnern, dass das Ziel der Strukturbiologie nicht darin besteht, die 3D-Struktur von Biomolekülen und ihren Komplexen vorherzusagen, sondern ihr Verhalten vorherzusagen und biologische Ergebnisse zu erzielen passiert, wenn Sie programmieren.

- Um Fortschritte bei der Vorhersage des molekularen Verhaltens zu erzielen, müssen wir uns darüber im Klaren sein, dass das Problem der Strukturvorhersage nicht so eindeutig ist, wie es scheint. Biomoleküle und ihre Komplexe falten sich nicht zu einer einzigen Struktur, sondern bilden stattdessen eine Ansammlung Tausender verschiedener Konformationen mit jeweils unterschiedlichen Wahrscheinlichkeiten und Lebensdauern.
- Das Verständnis dieser Konformationslandschaften und ihrer Veränderung bei der Interaktion von Biomolekülen ist für die quantitative Vorhersage von Affinitäten und kinetischen Raten von entscheidender Bedeutung.
- Die Vorhersage von Konformationsensembles aus Sequenzen unter verschiedenen Bedingungen ist ein Problem, auf das wir uns jetzt konzentrieren müssen, um ein quantitatives und prädiktives Verständnis des molekularen Verhaltens zu erlangen.
- Während die Verwendung von AlphaFold 3 zur Vorhersage der freien und komplexen 3D-Strukturen von Biomolekülen auf der Grundlage ihrer Sequenzen ein wichtiger Schritt zum Verständnis des molekularen Verhaltens und der Bioinformatik ist, müssen Experimentatoren keine Angst haben, zurückgelassen zu werden. Das Gebiet der Strukturbiologie wird noch dynamischer.
Link zum Papier: https://www.nature.com/articles/s41594-024-01350-2
Das obige ist der detaillierte Inhalt von„AlphaFold 3 macht einen wichtigen Schritt in Richtung Entschlüsselung molekularen Verhaltens und biologischer Datenverarbeitung', kommentierte ein „Nature'-Unterjournal. Für weitere Informationen folgen Sie bitte anderen verwandten Artikeln auf der PHP chinesischen Website!

Heiße KI -Werkzeuge

Undresser.AI Undress
KI-gestützte App zum Erstellen realistischer Aktfotos

AI Clothes Remover
Online-KI-Tool zum Entfernen von Kleidung aus Fotos.

Undress AI Tool
Ausziehbilder kostenlos

Clothoff.io
KI-Kleiderentferner

Video Face Swap
Tauschen Sie Gesichter in jedem Video mühelos mit unserem völlig kostenlosen KI-Gesichtstausch-Tool aus!

Heißer Artikel
Heiße Werkzeuge

Notepad++7.3.1
Einfach zu bedienender und kostenloser Code-Editor

SublimeText3 chinesische Version
Chinesische Version, sehr einfach zu bedienen

Senden Sie Studio 13.0.1
Leistungsstarke integrierte PHP-Entwicklungsumgebung

Dreamweaver CS6
Visuelle Webentwicklungstools

SublimeText3 Mac-Version
Codebearbeitungssoftware auf Gottesniveau (SublimeText3)
Heiße Themen
 1667
1667
 14
1426
52
1328
25
1273
29
1255
24
14
1426
52
1328
25
1273
29
1255
24

 „Defect Spectrum' durchbricht die Grenzen der herkömmlichen Fehlererkennung und erreicht erstmals eine hochpräzise und umfassende semantische Fehlererkennung in der Industrie.
Jul 26, 2024 pm 05:38 PM
„Defect Spectrum' durchbricht die Grenzen der herkömmlichen Fehlererkennung und erreicht erstmals eine hochpräzise und umfassende semantische Fehlererkennung in der Industrie.
Jul 26, 2024 pm 05:38 PM
In der modernen Fertigung ist die genaue Fehlererkennung nicht nur der Schlüssel zur Sicherstellung der Produktqualität, sondern auch der Kern für die Verbesserung der Produktionseffizienz. Allerdings mangelt es vorhandenen Datensätzen zur Fehlererkennung häufig an der Genauigkeit und dem semantischen Reichtum, die für praktische Anwendungen erforderlich sind, was dazu führt, dass Modelle bestimmte Fehlerkategorien oder -orte nicht identifizieren können. Um dieses Problem zu lösen, hat ein Spitzenforschungsteam bestehend aus der Hong Kong University of Science and Technology Guangzhou und Simou Technology innovativ den „DefectSpectrum“-Datensatz entwickelt, der eine detaillierte und semantisch reichhaltige groß angelegte Annotation von Industriedefekten ermöglicht. Wie in Tabelle 1 gezeigt, bietet der Datensatz „DefectSpectrum“ im Vergleich zu anderen Industriedatensätzen die meisten Fehleranmerkungen (5438 Fehlerproben) und die detaillierteste Fehlerklassifizierung (125 Fehlerkategorien).
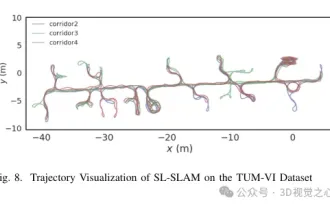 Jenseits von ORB-SLAM3! SL-SLAM: Szenen mit wenig Licht, starkem Jitter und schwacher Textur werden verarbeitet
May 30, 2024 am 09:35 AM
Jenseits von ORB-SLAM3! SL-SLAM: Szenen mit wenig Licht, starkem Jitter und schwacher Textur werden verarbeitet
May 30, 2024 am 09:35 AM
Heute diskutieren wir darüber, wie Deep-Learning-Technologie die Leistung von visionbasiertem SLAM (Simultaneous Localization and Mapping) in komplexen Umgebungen verbessern kann. Durch die Kombination von Methoden zur Tiefenmerkmalsextraktion und Tiefenanpassung stellen wir hier ein vielseitiges hybrides visuelles SLAM-System vor, das die Anpassung in anspruchsvollen Szenarien wie schlechten Lichtverhältnissen, dynamischer Beleuchtung, schwach strukturierten Bereichen und starkem Jitter verbessern soll. Unser System unterstützt mehrere Modi, einschließlich erweiterter Monokular-, Stereo-, Monokular-Trägheits- und Stereo-Trägheitskonfigurationen. Darüber hinaus wird analysiert, wie visuelles SLAM mit Deep-Learning-Methoden kombiniert werden kann, um andere Forschungen zu inspirieren. Durch umfangreiche Experimente mit öffentlichen Datensätzen und selbst abgetasteten Daten demonstrieren wir die Überlegenheit von SL-SLAM in Bezug auf Positionierungsgenauigkeit und Tracking-Robustheit.
 Training mit Millionen von Kristalldaten zur Lösung kristallographischer Phasenprobleme, die Deep-Learning-Methode PhAI wird in Science veröffentlicht
Aug 08, 2024 pm 09:22 PM
Training mit Millionen von Kristalldaten zur Lösung kristallographischer Phasenprobleme, die Deep-Learning-Methode PhAI wird in Science veröffentlicht
Aug 08, 2024 pm 09:22 PM
Herausgeber |KX Bis heute sind die durch die Kristallographie ermittelten Strukturdetails und Präzision, von einfachen Metallen bis hin zu großen Membranproteinen, mit keiner anderen Methode zu erreichen. Die größte Herausforderung, das sogenannte Phasenproblem, bleibt jedoch die Gewinnung von Phaseninformationen aus experimentell bestimmten Amplituden. Forscher der Universität Kopenhagen in Dänemark haben eine Deep-Learning-Methode namens PhAI entwickelt, um Kristallphasenprobleme zu lösen. Ein Deep-Learning-Neuronales Netzwerk, das mithilfe von Millionen künstlicher Kristallstrukturen und den entsprechenden synthetischen Beugungsdaten trainiert wird, kann genaue Elektronendichtekarten erstellen. Die Studie zeigt, dass diese Deep-Learning-basierte Ab-initio-Strukturlösungsmethode das Phasenproblem mit einer Auflösung von nur 2 Angström lösen kann, was nur 10 bis 20 % der bei atomarer Auflösung verfügbaren Daten im Vergleich zur herkömmlichen Ab-initio-Berechnung entspricht
 Das NVIDIA-Dialogmodell ChatQA wurde auf Version 2.0 weiterentwickelt, wobei die angegebene Kontextlänge 128 KB beträgt
Jul 26, 2024 am 08:40 AM
Das NVIDIA-Dialogmodell ChatQA wurde auf Version 2.0 weiterentwickelt, wobei die angegebene Kontextlänge 128 KB beträgt
Jul 26, 2024 am 08:40 AM
Die offene LLM-Community ist eine Ära, in der hundert Blumen blühen und konkurrieren. Sie können Llama-3-70B-Instruct, QWen2-72B-Instruct, Nemotron-4-340B-Instruct, Mixtral-8x22BInstruct-v0.1 und viele andere sehen hervorragende Darsteller. Allerdings weisen offene Modelle im Vergleich zu den proprietären Großmodellen GPT-4-Turbo in vielen Bereichen noch erhebliche Lücken auf. Zusätzlich zu allgemeinen Modellen wurden einige offene Modelle entwickelt, die sich auf Schlüsselbereiche spezialisieren, wie etwa DeepSeek-Coder-V2 für Programmierung und Mathematik und InternVL für visuelle Sprachaufgaben.
 Google AI gewann die Silbermedaille der IMO Mathematical Olympiad, das mathematische Argumentationsmodell AlphaProof wurde eingeführt und Reinforcement Learning ist zurück
Jul 26, 2024 pm 02:40 PM
Google AI gewann die Silbermedaille der IMO Mathematical Olympiad, das mathematische Argumentationsmodell AlphaProof wurde eingeführt und Reinforcement Learning ist zurück
Jul 26, 2024 pm 02:40 PM
Für KI ist die Mathematikolympiade kein Problem mehr. Am Donnerstag hat die künstliche Intelligenz von Google DeepMind eine Meisterleistung vollbracht: Sie nutzte KI, um meiner Meinung nach die eigentliche Frage der diesjährigen Internationalen Mathematikolympiade zu lösen, und war nur einen Schritt davon entfernt, die Goldmedaille zu gewinnen. Der IMO-Wettbewerb, der gerade letzte Woche zu Ende ging, hatte sechs Fragen zu Algebra, Kombinatorik, Geometrie und Zahlentheorie. Das von Google vorgeschlagene hybride KI-System beantwortete vier Fragen richtig und erzielte 28 Punkte und erreichte damit die Silbermedaillenstufe. Anfang dieses Monats hatte der UCLA-Professor Terence Tao gerade die KI-Mathematische Olympiade (AIMO Progress Award) mit einem Millionenpreis gefördert. Unerwarteterweise hatte sich das Niveau der KI-Problemlösung vor Juli auf dieses Niveau verbessert. Beantworten Sie die Fragen meiner Meinung nach gleichzeitig. Am schwierigsten ist es meiner Meinung nach, da sie die längste Geschichte, den größten Umfang und die negativsten Fragen haben
 PRO |. Warum verdienen große Modelle, die auf MoE basieren, mehr Aufmerksamkeit?
Aug 07, 2024 pm 07:08 PM
PRO |. Warum verdienen große Modelle, die auf MoE basieren, mehr Aufmerksamkeit?
Aug 07, 2024 pm 07:08 PM
Im Jahr 2023 entwickeln sich fast alle Bereiche der KI in beispielloser Geschwindigkeit weiter. Gleichzeitig verschiebt die KI ständig die technologischen Grenzen wichtiger Bereiche wie der verkörperten Intelligenz und des autonomen Fahrens. Wird der Status von Transformer als Mainstream-Architektur großer KI-Modelle durch den multimodalen Trend erschüttert? Warum ist die Erforschung großer Modelle auf Basis der MoE-Architektur (Mixture of Experts) zu einem neuen Trend in der Branche geworden? Können Large Vision Models (LVM) ein neuer Durchbruch im allgemeinen Sehvermögen sein? ...Aus dem PRO-Mitglieder-Newsletter 2023 dieser Website, der in den letzten sechs Monaten veröffentlicht wurde, haben wir 10 spezielle Interpretationen ausgewählt, die eine detaillierte Analyse der technologischen Trends und industriellen Veränderungen in den oben genannten Bereichen bieten, um Ihnen dabei zu helfen, Ihre Ziele in der Zukunft zu erreichen Jahr vorbereitet sein. Diese Interpretation stammt aus Week50 2023
 AlphaFold 3 wird auf den Markt gebracht und sagt die Wechselwirkungen und Strukturen von Proteinen und allen Lebensmolekülen umfassend und mit weitaus größerer Genauigkeit als je zuvor voraus
Jul 16, 2024 am 12:08 AM
AlphaFold 3 wird auf den Markt gebracht und sagt die Wechselwirkungen und Strukturen von Proteinen und allen Lebensmolekülen umfassend und mit weitaus größerer Genauigkeit als je zuvor voraus
Jul 16, 2024 am 12:08 AM
Herausgeber | Rettichhaut Seit der Veröffentlichung des leistungsstarken AlphaFold2 im Jahr 2021 verwenden Wissenschaftler Modelle zur Proteinstrukturvorhersage, um verschiedene Proteinstrukturen innerhalb von Zellen zu kartieren, Medikamente zu entdecken und eine „kosmische Karte“ jeder bekannten Proteininteraktion zu zeichnen. Gerade hat Google DeepMind das AlphaFold3-Modell veröffentlicht, das gemeinsame Strukturvorhersagen für Komplexe wie Proteine, Nukleinsäuren, kleine Moleküle, Ionen und modifizierte Reste durchführen kann. Die Genauigkeit von AlphaFold3 wurde im Vergleich zu vielen dedizierten Tools in der Vergangenheit (Protein-Ligand-Interaktion, Protein-Nukleinsäure-Interaktion, Antikörper-Antigen-Vorhersage) deutlich verbessert. Dies zeigt, dass dies innerhalb eines einzigen einheitlichen Deep-Learning-Frameworks möglich ist
 Um ein neues wissenschaftliches und komplexes Frage-Antwort-Benchmark- und Bewertungssystem für große Modelle bereitzustellen, haben UNSW, Argonne, die University of Chicago und andere Institutionen gemeinsam das SciQAG-Framework eingeführt
Jul 25, 2024 am 06:42 AM
Um ein neues wissenschaftliches und komplexes Frage-Antwort-Benchmark- und Bewertungssystem für große Modelle bereitzustellen, haben UNSW, Argonne, die University of Chicago und andere Institutionen gemeinsam das SciQAG-Framework eingeführt
Jul 25, 2024 am 06:42 AM
Herausgeber | Der Frage-Antwort-Datensatz (QA) von ScienceAI spielt eine entscheidende Rolle bei der Förderung der Forschung zur Verarbeitung natürlicher Sprache (NLP). Hochwertige QS-Datensätze können nicht nur zur Feinabstimmung von Modellen verwendet werden, sondern auch effektiv die Fähigkeiten großer Sprachmodelle (LLMs) bewerten, insbesondere die Fähigkeit, wissenschaftliche Erkenntnisse zu verstehen und zu begründen. Obwohl es derzeit viele wissenschaftliche QS-Datensätze aus den Bereichen Medizin, Chemie, Biologie und anderen Bereichen gibt, weisen diese Datensätze immer noch einige Mängel auf. Erstens ist das Datenformular relativ einfach, die meisten davon sind Multiple-Choice-Fragen. Sie sind leicht auszuwerten, schränken jedoch den Antwortauswahlbereich des Modells ein und können die Fähigkeit des Modells zur Beantwortung wissenschaftlicher Fragen nicht vollständig testen. Im Gegensatz dazu offene Fragen und Antworten
