


Herausgeber |. ScienceAI
Durch die Verwendung fortschrittlicher Sequenzmodelle wie Transformer wird das Problem der Vorhersage der einstufigen Retrosynthese in eine Übersetzungsaufgabe von der SMILES-Darstellung des Produkts zur SMILES-Darstellung des Reaktanten umgewandelt, was zu einer weit verbreiteten Strategie mit bemerkenswerten Ergebnissen geworden ist.
Allerdings lässt diese Methode oft einen entscheidenden Punkt außer Acht: Zwischen den Reaktanten und Produkten gibt es eine Vielzahl identischer Teilstrukturen, die direkt genutzt werden können. Eine unzureichende Nutzung dieser Unterstrukturen schränkt die Effizienz und Genauigkeit von Modellvorhersagen ein.
Im Juli 2024 veröffentlichte das Forschungsteam um Jin Yaohui und „Journal of Cheminformatics“. In der Studie schlug der Autor einen einstufigen retrosynthetischen Vorhersageprozess vor, der eine unbeaufsichtigte SMILES-Sequenzausrichtungstechnologie integriert, mit dem Ziel, die Genauigkeit und Effizienz der Vorhersage chemischer Reaktionen zu verbessern. Die experimentellen Ergebnisse belegen die Wirksamkeit des Modells bei der Vorhersage retrosynthetischer Wege und legen nahe, dass das Modell das Potenzial hat, ein wertvolles Werkzeug für die Arzneimittelentwicklung zu werden.
Link zum Papier: https://jcheminf.biomedcentral.com/articles/10.1186/s13321-024-00877-2
https://jcheminf.biomedcentral.com/articles/10.1186/s13321-024-00877-2
Wenn Atome als Knoten betrachtet werden, Durch die Behandlung chemischer Bindungen als Kanten kann die Molekülstruktur auf natürliche Weise in eine Graphenstruktur umgewandelt werden. Im Vergleich zu Sequenzmodellen können graphische neuronale Netze die topologischen Strukturinformationen innerhalb von Molekülen besser erfassen und so eine genauere molekulare Charakterisierung erreichen.
Darüber hinaus enthalten chemische Bindungen in chemischen Molekülen im Vergleich zu anderen Graphstrukturen umfangreiche Informationen zu chemischen Eigenschaften. Basierend auf diesen Vorteilen schlägt der Autor eine auf dem Graph Attention Network basierende Variante vor, um den Encoder-Teil im Transformer-Modell zu ersetzen, mit dem Ziel, leistungsfähigere molekulare Darstellungsmöglichkeiten für nachgelagerte Anwendungen bereitzustellen.Abbildung: Schematische Darstellung des Modells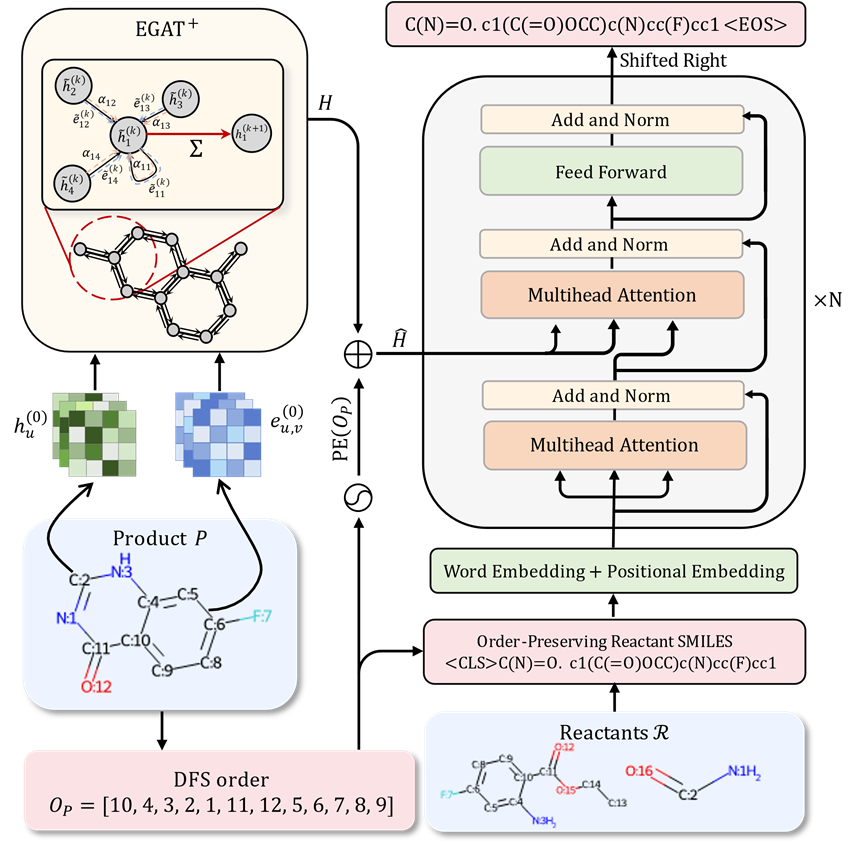
Bei der einstufigen Retrosynthesevorhersage bedeutet die Verwendung von Sequenzmodellierungsmethoden normalerweise, dass die Struktur der Reaktanten von Grund auf neu konstruiert werden muss Es ist nicht möglich, direkte Modifikationen auf der Grundlage vorhandener Produkte vorzunehmen, um identische Unterstrukturen zwischen Reaktanten und Produkten effizient zu nutzen. Dieser Ansatz schränkt die Genauigkeit der generierten Ergebnisse in gewissem Maße ein.
Wenn man bedenkt, dass die molekulare SMILES-Darstellung, die üblicherweise in der Sequenzmodellierung verwendet wird, die Atome und chemischen Bindungen im Molekül tatsächlich in der Reihenfolge der Tiefensuche anordnet, wenn die Positionsinformationen jedes Produktatoms, das in der Reaktanten-SMILES-Darstellung erscheint, bereitgestellt werden können Das Modell hilft dem Modell, Unterstrukturen zu identifizieren und wiederzuverwenden, die sich während der Reaktion nicht geändert haben. Dadurch wird die Schwierigkeit für das Modell, Reaktanten vorherzusagen, erheblich verringert und die Genauigkeit der Vorhersagen verbessert. Aus Sicht der Sequenzmodellierung ordnet die häufig verwendete molekulare SMILES-Charakterisierung die Atome und chemischen Bindungen im Molekül im Wesentlichen gemäß der Reihenfolge der Tiefensuche (DFS) an. Wenn dem Modell die Positionsinformationen jedes Atoms im Produkt in der SMILES-Darstellung der Reaktanten zur Verfügung gestellt werden können, wird dies die Identifizierung und Wiederverwendung unveränderter Unterstrukturen durch das Modell erheblich erleichtern, wodurch die Schwierigkeit der Vorhersage der Reaktanten erheblich verringert und die Genauigkeit verbessert wird . Die direkte Bereitstellung dieser Korrespondenzinformationen kann jedoch das Risiko eines Informationsverlusts während des Modelltrainings mit sich bringen. Um dieses Problem zu vermeiden, schlugen die Forscher eine innovative Strategie vor, um die Fähigkeit des Modells zu optimieren, die molekulare Struktur der Reaktanten zu verstehen und vorherzusagen, ohne Markierungsinformationen preiszugeben. Angesichts der Tatsache, dass die SMILES-Sequenzcharakterisierung aus einer Tiefensuche in Moleküldiagrammen abgeleitet wird und die meisten Unterstrukturen zwischen Reaktanten und Produkten hochgradig konsistent sind, muss es für eine gegebene DFS-Sequenz eines beliebigen Produkts eine entsprechende DFS-Reihenfolge auf dem Molekül geben Das Diagramm der Reaktanten ist so, dass die entsprechenden Atome auf den Reaktanten und Produkten in nahezu derselben Reihenfolge erscheinen. Basierend auf dieser Strategie haben die Forscher nicht nur die Molekülstruktur des Produkts in die Modelleingabe einbezogen, sondern auch die DFS-Reihenfolge der Reaktantenmoleküle als Teil der Eingabe eingeführt. Darüber hinaus generierten die Forscher gemäß der oben genannten Strategie eine Produktmolekül-DFS-Sequenz, die in hohem Maße mit der DFS-Sequenz eines bestimmten Reaktanten übereinstimmt, und verwendeten diese Sequenz, um eine SMILES-Darstellung des Reaktanten als Ziel des Modelltrainings zu generieren . Dieses Design ermöglicht die Anordnung ähnlicher Unterstrukturen zwischen Reaktanten und Produkten in nahezu der gleichen Reihenfolge in der Eingabe und Ausgabe des Modells, wodurch der Prozess des Modelllernens der gleichen strukturellen Entsprechung zwischen Reaktanten und Produkten vereinfacht und die Identifizierung der Gruppen erleichtert wird die sich während der Reaktion ändern.Selbst wenn die Reaktantenstruktur von Grund auf neu erstellt wird, kann diese Methode Produktstrukturinformationen effektiv wiederverwenden und die Genauigkeit der Vorhersage erheblich verbessern.
Besonders wichtig ist, dass diese Methode das Problem des Label-Lecks während des Modelltrainingsprozesses effektiv vermeidet, da die DFS-Reihenfolge des Produkts nur auf seinen molekularen Strukturinformationen basiert und nicht auf Informationen über die Reaktanten als Annotationen beruht.
Gleichzeitig erfordert diese unbeaufsichtigte SMILES-Ausrichtungsmethode nicht die Einführung zusätzlicher Überwachungssignale während des Trainingsprozesses, wodurch komplexe Datenannotations- und Optimierungsprobleme beim Lernen mit mehreren Aufgaben vermieden werden und eine neuartige Methode für den Bereich der Molekularbiologie bereitgestellt wird Retrosynthesevorhersage und effiziente Forschungsmethoden.
Anzeige der experimentellen Ergebnisse
In dieser Studie führte der Autor eine systematische Auswertung mehrerer Datensätze zur Vorhersage der molekularen Retrosynthese durch und deckte dabei den weit verbreiteten USPTO-50K-Datensatz sowie den USPTO-50K-Datensatz mit einer größeren Menge ab von Daten. MIT und USPTO-FULL.
Bei der Bewertung der Modellleistung wird die Top-K-Genauigkeit als Hauptbewertungsindex verwendet. Am USPTO-50K-Datensatz untersuchte der Autor nicht nur die Rechtmäßigkeit der vom Modell generierten SMILES-Sequenz, sondern führte auch eine Loopback-Überprüfung der praktischen Machbarkeit des vom Modell ausgegebenen Syntheseschemas durch ein groß angelegtes vorab trainiertes Modell durch Modell zur Vorhersage der Vorwärtsreaktion.
Tabelle 1: Höchste Genauigkeit der USPTO-50K-Retrosynthesevorhersagen
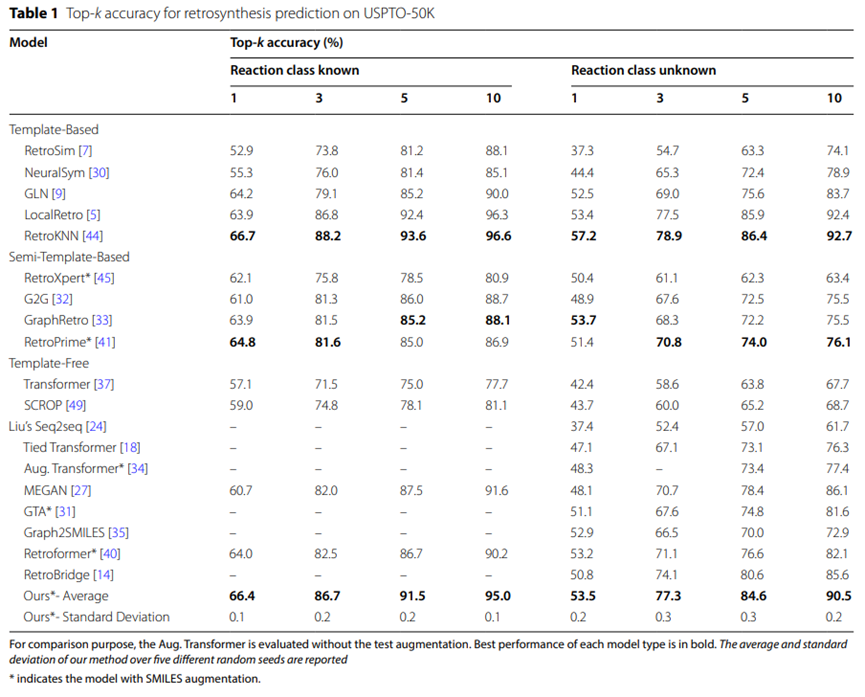
Die experimentellen Ergebnisse des USPTO-50K-Datensatzes sind in Tabelle 1 zusammengefasst und zeigen, dass das UAlign-Modell in USPTO eine bessere Leistung erbringt, wenn der spezifische Reaktionstyp vorliegt ist nicht angegeben Die Top-5-Genauigkeit des -50K-Datensatzes beträgt bis zu 84,6 % und ist damit deutlich besser als bei anderen vorlagenfreien Basismodellen.
Tabelle 2: Top-k-Genauigkeit der retrosynthetischen USPTO-MIT-Vorhersage
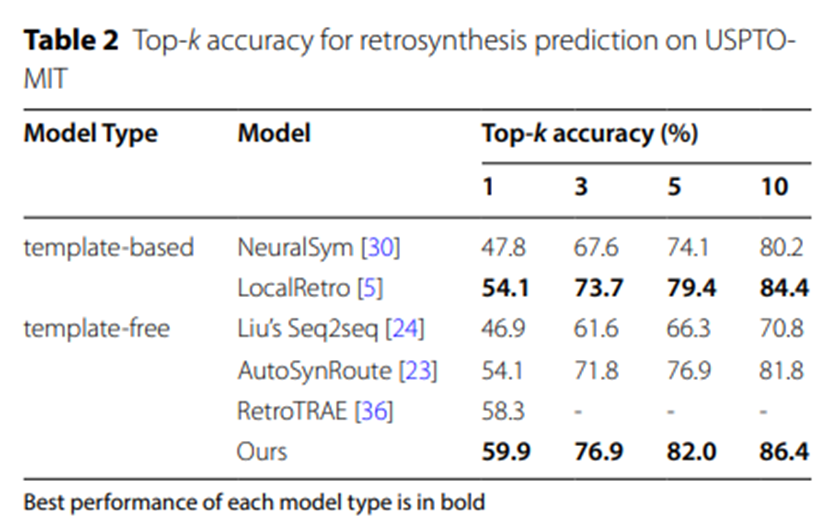
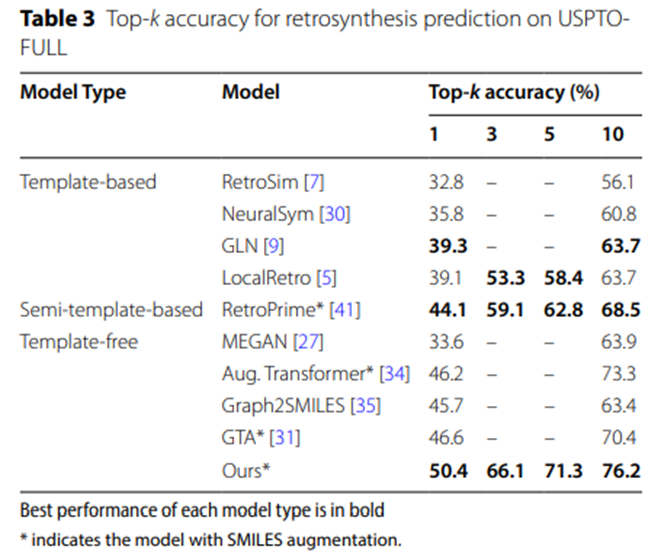
Die experimentellen Daten in Tabelle 2 und Tabelle 3 bestätigen weiterhin, dass bei den größeren Datensätzen USPTO-MIT und USPTO-FULL das UAlign-Modell verwendet wird übertrifft andere verschiedene Basismodelle durch erhebliche Vorteile.
Tabelle 3: Top-k-Genauigkeit der retrosynthetischen Vorhersage auf USPTO-FULL
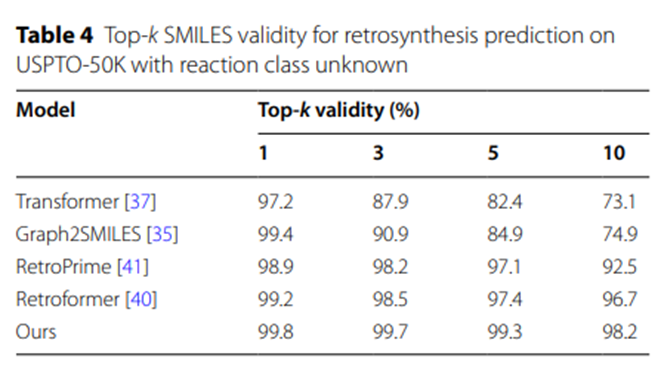
Darüber hinaus zeigen die experimentellen Ergebnisse in Tabelle 4, dass im Vergleich zu anderen SMILES-basierten retrosynthetischen Vorhersagemodellen die vom UAlign-Modell erzeugten Reaktanten The Die SMILES-Sequenz hat eine höhere Legitimität.
Tabelle 4: Top-k-SMILES-Wirksamkeit für retrosynthetische Vorhersagen unbekannter Reaktionsklassen auf USPTO-50K
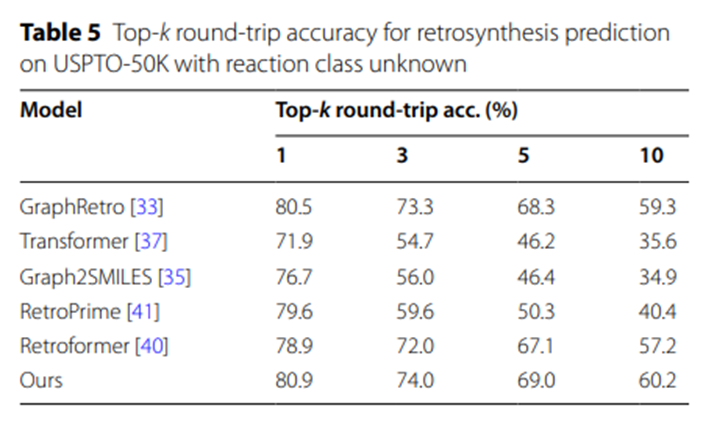
Die experimentellen Daten in Tabelle 5 unterstreichen zusätzlich die Fähigkeit des UAlign-Modells, vernünftige und realisierbare Syntheseschemata zu generieren. Der Grund dafür ist, dass ein relativ großer Teil der von UAlign vorgeschlagenen Syntheseschemata die Verifizierung des Vorwärtsreaktionsvorhersagemodells bestehen kann, d. h. diese Schemata können nach entsprechenden chemischen Reaktionen effektiv in bestimmte Zielprodukte umgewandelt werden.
Tabelle 5: Top-k-Round-Trip-Genauigkeit für die Retrosynthesevorhersage mit unbekannten Reaktionskategorien auf USPTO-50K
Diese experimentellen Ergebnisse bestätigen nicht nur die Effizienz und Genauigkeit des UAlign-Modells bei der Vorhersageaufgabe der molekularen Retrosynthese, sondern auch Außerdem unterstreicht es seine hervorragende Leistung bei der Verarbeitung großer Datensätze und seine erheblichen Vorteile bei der Generierung hochwertiger Syntheselösungen.
Um das Anwendungspotenzial des UAlign-Modells in der tatsächlichen Produktion zu überprüfen, wählte der Autor in den letzten zwei Jahren neue Medikamente aus, die von der US-amerikanischen Food and Drug Administration (FDA) als Syntheseziele zugelassen wurden Die Synthese wurde erfolgreich durchgeführt. Die Vorhersagen des Modells zu den Synthesewegen dieser beiden Medikamente stimmen in hohem Maße mit den in der Literatur dokumentierten Wegen überein.
Darüber hinaus wurde auch für das dritte Medikament der vom Modell vorhergesagte Syntheseweg von Experten auf dem Gebiet der Chemie als machbar anerkannt. Diese Synthesewege decken nicht nur eine Vielzahl von Reaktionstypen ab, sondern umfassen auch komplexe Situationen wie die Synthese zyklischer Verbindungen und einstufige Retrosynthesevorhersagen, an denen mehrere Reaktionszentren beteiligt sind.
Die oben genannten experimentellen Ergebnisse beweisen voll und ganz, dass das UAlign-Modell nicht nur verschiedene Reaktionstypen bewältigen kann, sondern auch einen hohen Anwendungswert in der tatsächlichen Produktion hat. Dies zeigt, dass das UAlign-Modell eine hohe Praktikabilität und Flexibilität im Bereich der Vorhersage der molekularen Retrosynthese aufweist und effektive Lösungen für die Arzneimittelsynthese bieten kann.

Zukunftsausblick
Mit seiner hervorragenden Leistung und Flexibilität ist das UAlign-Modell durchaus in der Lage, als Eckpfeiler für den Aufbau eines mehrstufigen Retrosynthesesystems zu dienen. Es kann mit verschiedenen Suchalgorithmen und Multi-Ziel-Optimierungstechnologie kombiniert werden, um ein effizientes und intelligentes retrosynthetisches Pfadplanungssystem zu bilden.
Darüber hinaus erforscht der Autor aktiv die Integration des UAlign-Algorithmus mit fortschrittlicher Hardwareausrüstung, um ein automatisiertes unbemanntes Labor zu schaffen, das die Automatisierung von Arzneimittelentdeckungs- und -syntheseprozessen vorantreibt und revolutionäre Veränderungen in den Bereichen der chemischen Forschung und Arzneimittelentwicklung mit sich bringt. ändern.
Das obige ist der detaillierte Inhalt vonChemische Retrosynthese SOTA! Das Team der Shanghai Jiao Tong University schlägt die SMILES-Ausrichtungstechnologie vor, um eine effiziente retrosynthetische Vorhersage zu erreichen. Für weitere Informationen folgen Sie bitte anderen verwandten Artikeln auf der PHP chinesischen Website!
 So öffnen Sie HTML-Dateien
So öffnen Sie HTML-Dateien
 Tutorial zur Linux-Systeminstallation
Tutorial zur Linux-Systeminstallation
 Analyse der ICP-Münzaussichten
Analyse der ICP-Münzaussichten
 Thunder VIP-Patch
Thunder VIP-Patch
 Fehlerberichtslösung für den MySQL-Import einer SQL-Datei
Fehlerberichtslösung für den MySQL-Import einer SQL-Datei
 So lösen Sie das Problem, wenn der Computer eingeschaltet wird, der Bildschirm schwarz wird und der Desktop nicht aufgerufen werden kann
So lösen Sie das Problem, wenn der Computer eingeschaltet wird, der Bildschirm schwarz wird und der Desktop nicht aufgerufen werden kann
 Einführung in die Verwendung von vscode
Einführung in die Verwendung von vscode
 Virtuelle Währungsumtauschplattform
Virtuelle Währungsumtauschplattform








![[Web-Frontend] Node.js-Schnellstart](https://img.php.cn/upload/course/000/000/067/662b5d34ba7c0227.png)



