


Editor |. KX
Das von Google DeepMind-Forschern entwickelte Fermion-Neuronale Netzwerk (FermiNet) eignet sich sehr gut zur Modellierung des Quantengrundzustands einer großen Anzahl von Elektronen.
FermiNet konzentrierte sich zunächst auf den Grundzustand von Molekülen. Wenn Moleküle und Materialien jedoch durch große Energiemengen angeregt werden, beispielsweise wenn sie Licht oder hohen Temperaturen ausgesetzt werden, können Elektronen in einen Zustand höherer Energie versetzt werden – einen angeregten Zustand.
Angeregte Zustände sind in Bereichen wie Physik und Chemie wichtig; skalierbare, genaue und robuste Berechnungen angeregter Zustandseigenschaften auf der Grundlage erster Prinzipien stehen jedoch immer noch vor großen theoretischen Herausforderungen.
Jetzt haben DeepMind-Forscher eine neue Methode zur Berechnung angeregter Zustände entwickelt, die leistungsfähiger und vielseitiger ist als frühere Methoden. Die Methode kann auf jede Art von mathematischem Modell angewendet werden, einschließlich FermiNet und anderen neuronalen Netzen.
Die vorgeschlagene Methode ermöglicht genaue Berechnungen des angeregten Zustands für viele Atome und Moleküle, ist bestehenden Methoden zur Verwendung von Deep Learning zur Berechnung der Eigenschaften angeregter Zustände (insbesondere bei größeren Systemen) weit überlegen und kann auf verschiedene Quantensysteme angewendet werden.
David Pfau, der Erstautor und korrespondierende Autor des Papiers, veröffentlichte begeistert: „Dies ist das erste Mal, dass Deep Learning einige der schwierigsten Probleme der Quantenphysik genau gelöst hat. Wir hoffen, einen neuen Schritt in Richtung universeller Quantensimulation zu machen.“ des Deep Learning.“

Relevante Forschungsergebnisse mit dem Titel „Genaue Berechnung quantenangeregter Zustände mit neuronalen Netzen“ wurden auf Science veröffentlicht!
Link zum Papier: https://www.science.org/doi/abs/10.1126/science.adn0137
Molekulare angeregte Zustände
Wenn Moleküle und Materialien durch große Energiemengen stimuliert werden, Wenn sie beispielsweise Licht oder hohen Temperaturen ausgesetzt werden, gelangen ihre Elektronen in eine vorübergehende neue Struktur, die als angeregter Zustand bezeichnet wird.
Die genaue Energiemenge, die ein Molekül beim Übergang zwischen Zuständen absorbiert und freisetzt, erzeugt einen einzigartigen Fingerabdruck für verschiedene Moleküle und Materialien. Dies wirkt sich auf die Leistung von Technologien aus, die von Solarmodulen und LEDs bis hin zu Halbleitern und Photokatalysatoren reichen. Sie spielen auch eine Schlüsselrolle bei biologischen Prozessen, an denen Licht beteiligt ist, einschließlich der Photosynthese und des Sehens.
Allerdings ist diese Art von Fingerabdruck äußerst schwierig zu modellieren, da die angeregten Elektronen Quantennatur haben, was bedeutet, dass ihre Position im Molekül niemals sicher ist und nur durch Wahrscheinlichkeiten dargestellt werden kann.
FermiNet kann hochpräzise und in einigen Fällen hochmoderne Grundzustandsenergien in einer Reihe von Atomen und kleinen Molekülen mit einer Vielzahl qualitativer Positronenbindungseigenschaften erzeugen.
Allerdings konzentrierte sich FermiNet zunächst auf den Grundzustand von Molekülen. Wenn Moleküle und Materialien jedoch durch große Energiemengen angeregt werden, beispielsweise wenn sie Licht oder hohen Temperaturen ausgesetzt werden, können Elektronen in einen Zustand höherer Energie versetzt werden – einen angeregten Zustand.
Die genaue Berechnung der Energie des angeregten Zustands ist viel schwieriger als die Berechnung der Energie des Grundzustands. Sogar Goldstandardmethoden der Grundzustandschemie, wie etwa gekoppelte Cluster, zeigen zehnfache Fehler in angeregten Zuständen. Während die Forscher ihre Arbeit an FermiNet auf angeregte Zustände ausweiten wollten, waren die vorhandenen Methoden nicht leistungsfähig genug, um neuronale Netze mit modernsten Methoden vergleichbar zu machen.
Eine leistungsfähigere und vielseitigere neue Methode zur Berechnung angeregter Zustände
DeepMind schlägt einen Algorithmus zur Schätzung angeregter Zustände von Quantensystemen durch Variations-Monte-Carlo vor. Dieser Algorithmus hat keine freien Parameter und erfordert keine Orthogonalisierung der Zustände Transformieren Sie das Problem in das Problem, den Grundzustand des erweiterten Systems zu finden. Es können beliebige Observablen berechnet werden, einschließlich nichtdiagonaler Erwartungen wie Übergangsdipolmomente.
Diese Methode eignet sich besonders für die Analyse neuronaler Netzwerke. Durch die Kombination dieser Methode mit dem FermiNet- und Psiformer-Ansatz ist es möglich, die Anregungsenergien und Oszillatorstärken einer Reihe von Molekülen genau wiederherzustellen.

Abbildung: Angeregte Zustandsenergien der ersten Atomreihe von Lithium bis Neon. Ergebnisse von NES-VMC angewendet auf FermiNet. (Quelle: Papier)
Die Forscher kombinierten die Flexibilität neuronaler Netzansätze mit mathematischen Erkenntnissen und ermöglichten es ihnen, das Problem, den angeregten Zustand eines Systems zu finden, in das Problem, den Grundzustand eines erweiterten Systems zu finden, umzuwandeln, das dann kann mit Standard-VMC gelöst werden. Diese Methode wird als natürlich angeregter Zustand VMC (NES-VMC) bezeichnet.
Die lineare Unabhängigkeit angeregter Zustände wird automatisch über die funktionale Form des Ansatzes erzwungen. Die Energie jedes angeregten Zustands und anderer Observablen werden durch Diagonalisierung der Hamiltonschen Erwartungswertmatrix auf den Singulett-Ansätzen erhalten, und diese Observablen können ohne zusätzliche Kosten akkumuliert werden.
Entscheidend ist, dass diese Methode keine freien Parameter zum Anpassen und keinen Strafterm zum Erzwingen der Orthogonalisierung hat. Die Forscher untersuchten die Genauigkeit dieses Ansatzes mithilfe zweier unterschiedlicher neuronaler Netzwerkarchitekturen: FermiNet und Psiformer.
Von einzelnen Atomen zu Benzol
Die Forscher testeten ihre Methode an einem Benchmark-System, das von einzelnen Atomen bis zu Molekülen in Benzolgröße reicht. Es wurde bestätigt, dass die Genauigkeit von NES-VMC bei Atomen der ersten Reihe den experimentellen Ergebnissen sehr nahe kommt, und bei einer Reihe kleiner Moleküle werden hochpräzise Energien und Schwingungen erhalten, die mit den besten vorhandenen theoretischen Gerätestärken vergleichbar sind.
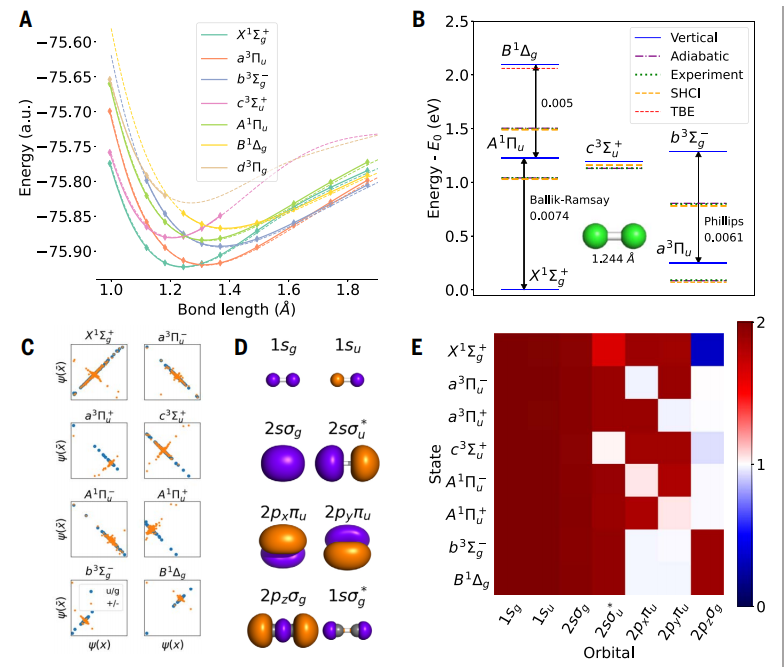
Abbildung: angeregter Zustand des Kohlenstoffdimers. (Quelle: Paper)
Erzielte einen mittleren absoluten Fehler (MAE) von 4 meV bei einem kleinen, komplexen Molekül namens Kohlenstoffdimer, was fünfmal genauer ist als frühere Goldstandardberechnungen.

Abbildung: angeregte Zustände und konische Schnittpunkte von Ethylen. (Quelle: Papier)
Im Fall von Ethylen beschreibt NES-VMC die konischen Schnittpunkte des verdrillten Moleküls korrekt und stimmt gut mit den hochpräzisen Ergebnissen der Multiple Reference Configuration Interaction (MR-CI) überein.

Abbildung: Angeregte Zustände eines größeren Doppelerregungssystems. (Quelle: Papier)
Die Studie berücksichtigte auch fünf anspruchsvolle Systeme mit tiefliegenden Doppelanregungen, darunter mehrere Moleküle der Benzolklasse. In einem System, in dem alle Methoden eine gute Übereinstimmung hinsichtlich der vertikalen Anregungsenergien aufweisen, ist Psiformer in allen Zuständen chemisch genau, einschließlich Butadien, dessen Reihenfolge bei einigen ebenfalls seit Jahrzehnten umstritten ist.
Für Tetrazine und Cyclopentadienone, bei denen hochmoderne Berechnungen von vor einigen Jahren bekanntermaßen ungenau sind, stimmen die NES-VMC-Ergebnisse nicht mit der jüngsten hochentwickelten Monte-Carlo-Diffusion (DMC) und vollständig aktiven Raumstörung dritter Ordnung überein Die Berechnung der Theorien (CASPT3) liegt sehr nahe.

Abbildung: angeregter Zustand von Benzol. (Quelle: Papier)
Schließlich wurde auch das Benzolmolekül untersucht, wobei NES-VMC in Kombination mit dem Psiformer-Ansatz im Vergleich zu anderen Methoden, einschließlich des neuronalen Netzwerkansatzes unter Verwendung der Strafkonsistenz, bessere Ergebnisse lieferte. Dies bestätigt nicht nur die mathematische Korrektheit der vorgeschlagenen Methode, sondern zeigt auch, dass neuronale Netze die angeregten Zustände von Molekülen an den Grenzen aktueller Rechenmethoden genau darstellen können.
Wie man es in Zukunft auf die Vielteilchen-Quantenmechanik anwenden kann
NES-VMC ist ein parameterfreies und mathematisch fundiertes Prinzip der Variation angeregter Zustände. Kombiniert man dies mit neuronalen Netzwerkansätzen, kann bei einer Vielzahl von Benchmark-Problemen eine erhebliche Genauigkeit erreicht werden.
Genaue VMC-Methoden für angeregte Zustände von Quantensystemen eröffnen viele Möglichkeiten und erweitern den Anwendungsbereich neuronaler Netzwerkwellenfunktionen erheblich.
Obwohl diese Studie nur elektronische Anregungen molekularer Systeme und neuronale Netzwerkansätze berücksichtigte, ist NES-VMC auf jeden Quanten-Hamiltonianer und jeden Ansatz anwendbar und ermöglicht präzise rechnerische Studien, die das Verständnis der Wissenschaftler für die elektronische Schwingungskopplung und das optische Bandverständnis verbessern können Lücken, Kernphysik und andere herausfordernde Probleme.
Die Forscher sagten: „Wir freuen uns darauf, wie NES-VMC und tiefe neuronale Netze in Zukunft auf die anspruchsvollsten Probleme der Vielteilchen-Quantenmechanik angewendet werden.“ /x.com/pfau/status/1826681648597135464
https://deepmind.google/discover/blog/ferminet-quantum-physics-and-chemistry-from-first-principles/
https:// www.imperial .ac.uk/news/255673/ai-tackles-most-difficult-challenges-quantum/
Das obige ist der detaillierte Inhalt vonKI löst erstmals quantenphysikalisches Problem, DeepMind berechnet präzise angeregte Quantenzustände, aufgeführt in Science. Für weitere Informationen folgen Sie bitte anderen verwandten Artikeln auf der PHP chinesischen Website!
 Was sind die Hauptfunktionen von Redis?
Was sind die Hauptfunktionen von Redis?
 xrp Ripple Neueste Nachrichten
xrp Ripple Neueste Nachrichten
 So öffnen Sie JAR-Dateien
So öffnen Sie JAR-Dateien
 Wie man auf Douyin einen kleinen Feuerwehrmann großzieht
Wie man auf Douyin einen kleinen Feuerwehrmann großzieht
 Was soll ich tun, wenn die sekundäre Webseite nicht geöffnet werden kann?
Was soll ich tun, wenn die sekundäre Webseite nicht geöffnet werden kann?
 Was sind die Unterschiede zwischen C++ und der C-Sprache?
Was sind die Unterschiede zwischen C++ und der C-Sprache?
 Ändern Sie die Hintergrundfarbe des Wortes in Weiß
Ändern Sie die Hintergrundfarbe des Wortes in Weiß
 ntuser.dat hat die Wiederherstellungsmethode gelöscht
ntuser.dat hat die Wiederherstellungsmethode gelöscht








![[Web-Frontend] Node.js-Schnellstart](https://img.php.cn/upload/course/000/000/067/662b5d34ba7c0227.png)



