
 Technologie-Peripheriegeräte
KI
Schneller als 0! Meta hat ein großes Proteinmodell mit 15 Milliarden Parametern gestartet, um AlphaFold2 zu zerstören
Technologie-Peripheriegeräte
KI
Schneller als 0! Meta hat ein großes Proteinmodell mit 15 Milliarden Parametern gestartet, um AlphaFold2 zu zerstören
Schneller als 0! Meta hat ein großes Proteinmodell mit 15 Milliarden Parametern gestartet, um AlphaFold2 zu zerstören

Das bisher größte Protein-Sprachmodell wurde veröffentlicht!
Vor einem Jahr wurde DeepMinds Open-Source-Programm AlphaFold2 in den Bereichen Natur und Wissenschaft eingeführt und überwältigte die akademischen Kreise der Biologie und KI.
Ein Jahr später kam Meta mit ESMFold, das eine Größenordnung schneller war.
Es ist nicht nur schnell, das Modell verfügt auch über 15 Milliarden Parameter.

LeCun twitterte, um zu loben, dass dies eine großartige neue Errungenschaft des Meta-FAIR-Proteinteams sei.

Co-Autor Zeming Lin enthüllte, dass das große Modell mit 3 Milliarden Parametern 3 Wochen lang auf 256 GPUs trainiert wurde, während ESMfold 10 Tage auf 128 GPUs benötigte. Was die 15-Milliarden-Parameter-Version betrifft, ist noch unklar.
Er sagte auch, dass der Code später definitiv Open Source sein wird, also bleiben Sie dran!
Groß und schnell!
Heute ist unser Protagonist ESMFold, ein Modell, das hochgenaue End-to-End-Strukturen auf atomarer Ebene aus einzelnen Proteinsequenzen direkt vorhersagt.

Papieradresse: https://www.biorxiv.org/content/10.1101/2022.07.20.500902v1
Unnötig zu erwähnen, welche Vorteile 15 Milliarden Parameter mit sich bringen – durch Training können die heutigen großen Modelle die drei vorhersagen -dimensionale Struktur von Proteinen mit atomarer Größengenauigkeit.
In Bezug auf die Genauigkeit ist ESMFold fast identisch mit AlphaFold2 und RoseTTAFold.
Allerdings ist die Inferenzgeschwindigkeit von ESMFold um eine Größenordnung schneller als bei AlphaFold2!
Es kann schwierig sein, den Geschwindigkeitsvergleich zwischen den dreien zu verstehen, wenn man über die Größenordnung spricht. Schauen Sie sich einfach das Bild unten an, um es zu verstehen.

Was ist der Unterschied?
Obwohl AlphaFold2 und RoseTTAFold beim Problem der Strukturvorhersage mit atomarer Auflösung bahnbrechende Erfolge erzielt haben, verlassen sie sich auch auf die Verwendung mehrerer Sequenzalignments (MSA) und ähnlicher Proteinstrukturvorlagen, um eine optimale Leistung zu erzielen.
Im Gegensatz dazu kann ESMFold durch die Nutzung der internen Darstellung des Sprachmodells entsprechende Strukturvorhersagen mit nur einer Sequenz als Eingabe generieren und so die Strukturvorhersage erheblich beschleunigen.

Die Forscher fanden heraus, dass die Vorhersagen von ESMFold für Sequenzen mit geringer Komplexität mit aktuellen Modellen auf dem neuesten Stand der Technik vergleichbar waren.
Darüber hinaus hängt die Genauigkeit der Strukturvorhersage eng mit der Komplexität des Sprachmodells zusammen. Das heißt, wenn das Sprachmodell die Sequenz besser verstehen kann, kann es auch die Struktur besser verstehen.

Derzeit gibt es Milliarden von Proteinsequenzen unbekannter Struktur und Funktion, von denen viele aus der metagenomischen Sequenzierung stammen.
Mit ESMFold können Forscher eine Zufallsstichprobe von 1 Million metagenomischen Sequenzen in nur 6 Stunden falten.

Ein großer Teil davon weist ein hohes Vertrauen auf und unterscheidet sich von allen bekannten Strukturen (nicht in der Datenbank erfasst).
Forscher glauben, dass ESMFold dabei helfen kann, Proteinstrukturen zu verstehen, die jenseits des derzeitigen Verständnisses liegen.

Da die Vorhersagen von ESMFold um eine Größenordnung schneller sind als bestehende Modelle, können Forscher ESMFold außerdem dazu nutzen, die Lücke zwischen den schnell wachsenden Proteinsequenzdatenbanken und den langsamer wachsenden Proteinstruktur- und -funktionsdatenbanken zu schließen.
15-Milliarden-Parameter-Protein-Sprachmodell
Als nächstes sprechen wir ausführlich über Metas neues ESMFold.
ESM-2 ist ein Transformer-basiertes Sprachmodell und nutzt einen Aufmerksamkeitsmechanismus, um die Interaktionsmuster zwischen Aminosäurepaaren in der Eingabesequenz zu lernen.
Im Vergleich zum Vorgängermodell ESM-1b hat Meta die Modellstruktur und Trainingsparameter verbessert sowie Rechenressourcen und Daten hinzugefügt. Gleichzeitig ermöglicht die Hinzufügung der relativen Positionseinbettung die Verallgemeinerung des Modells auf Sequenzen beliebiger Länge.
Aus den Ergebnissen geht hervor, dass das ESM-2-Modell mit 150 Millionen Parametern eine bessere Leistung erbringt als das ESM-1b-Modell mit 650 Millionen Parametern.
Darüber hinaus übertraf ESM-2 auch andere Proteinsprachenmodelle beim Benchmark der Strukturvorhersage. Diese Leistungsverbesserung steht im Einklang mit etablierten Mustern im Bereich der groß angelegten Sprachmodellierung.

Mit zunehmendem Maßstab von ESM-2 kann beobachtet werden, dass sich die Genauigkeit der Sprachmodellierung erheblich verbessert hat.

End-to-End-Vorhersage der Einzelsequenzstruktur
Ein wesentlicher Unterschied zwischen SMFold und AlphaFold2 besteht darin, dass ESMFold eine Sprache verwendet Das darzustellende Modell macht eindeutige homologe Sequenzen (in Form von MSA) als Eingabe überflüssig.
ESMFold vereinfacht den Evoformer in AlphaFold2, indem es das rechenintensive Netzwerkmodul, das MSA verarbeitet, durch ein Transformer-Modul ersetzt, das Sequenzen verarbeitet. Diese Vereinfachung bedeutet, dass ESMFold deutlich schneller ist als MSA-basierte Modelle.
Die Ausgabe des gefalteten Rückgrats wird dann von einem Strukturmodul verarbeitet, das für die Ausgabe der endgültigen Struktur auf atomarer Ebene und der Vorhersagesicherheit verantwortlich ist.

Forscher testeten ESMFold mit AlphaFold2 und RoseTTAFold bei CAMEO (April 2022 bis Juni 2022) und CASP14 (Mai 2020) Vergleich am Set.
Wenn nur eine einzelne Sequenz als Eingabe angegeben wird, schneidet ESMFold viel besser ab als Alphafold 2.
Bei Nutzung der vollständigen Pipeline erreichte AlphaFold2 88,3 bzw. 84,7 auf CAMEO und CASP14. ESMFold erreicht auf CAMEO eine vergleichbare Genauigkeit wie RoseTTAfold mit einem durchschnittlichen TM-Score von 82,0. Die Forscher fanden heraus, dass Sprachmodelle, die auf unbeaufsichtigtes Lernen abzielen, auf einer großen Datenbank evolutionär unterschiedlicher Proteinsequenzen trainiert werden und in der Lage sind, Proteinstrukturen mit atomarer Auflösung vorherzusagen.
Durch die Erweiterung der Parameter des Sprachmodells auf 15B kann der Einfluss der Skalierung auf das Lernen der Proteinstruktur systematisch untersucht werden. 
Seiner persönlichen Homepage zufolge hat Zeming an der New York University promoviert und als Forschungsingenieur (Gast) bei Meta AI gearbeitet, wo er hauptsächlich für die Arbeit an der Back-End-Infrastruktur verantwortlich war.
Er studierte sowohl seinen Bachelor- als auch seinen Masterabschluss an der University of Virginia, wo er zusammen mit Yanjun Qi an Anwendungen des maschinellen Lernens, insbesondere zur Vorhersage der Proteinstruktur, forschte.
Die Interessengebiete sind Deep Learning, Strukturvorhersage und Informationsbiologie.
Das obige ist der detaillierte Inhalt vonSchneller als 0! Meta hat ein großes Proteinmodell mit 15 Milliarden Parametern gestartet, um AlphaFold2 zu zerstören. Für weitere Informationen folgen Sie bitte anderen verwandten Artikeln auf der PHP chinesischen Website!

Heiße KI -Werkzeuge

Undresser.AI Undress
KI-gestützte App zum Erstellen realistischer Aktfotos

AI Clothes Remover
Online-KI-Tool zum Entfernen von Kleidung aus Fotos.

Undress AI Tool
Ausziehbilder kostenlos

Clothoff.io
KI-Kleiderentferner

AI Hentai Generator
Erstellen Sie kostenlos Ai Hentai.

Heißer Artikel
Heiße Werkzeuge

Notepad++7.3.1
Einfach zu bedienender und kostenloser Code-Editor

SublimeText3 chinesische Version
Chinesische Version, sehr einfach zu bedienen

Senden Sie Studio 13.0.1
Leistungsstarke integrierte PHP-Entwicklungsumgebung

Dreamweaver CS6
Visuelle Webentwicklungstools

SublimeText3 Mac-Version
Codebearbeitungssoftware auf Gottesniveau (SublimeText3)
Heiße Themen
 1377
1377
 52
52

 Der DeepMind-Roboter spielt Tischtennis und seine Vor- und Rückhand rutschen in die Luft, wodurch menschliche Anfänger völlig besiegt werden
Aug 09, 2024 pm 04:01 PM
Der DeepMind-Roboter spielt Tischtennis und seine Vor- und Rückhand rutschen in die Luft, wodurch menschliche Anfänger völlig besiegt werden
Aug 09, 2024 pm 04:01 PM
Aber vielleicht kann er den alten Mann im Park nicht besiegen? Die Olympischen Spiele in Paris sind in vollem Gange und Tischtennis hat viel Aufmerksamkeit erregt. Gleichzeitig haben Roboter auch beim Tischtennisspielen neue Durchbrüche erzielt. Gerade hat DeepMind den ersten lernenden Roboteragenten vorgeschlagen, der das Niveau menschlicher Amateurspieler im Tischtennis-Wettkampf erreichen kann. Papieradresse: https://arxiv.org/pdf/2408.03906 Wie gut ist der DeepMind-Roboter beim Tischtennisspielen? Vermutlich auf Augenhöhe mit menschlichen Amateurspielern: Sowohl Vorhand als auch Rückhand: Der Gegner nutzt unterschiedliche Spielstile, und auch der Roboter hält aus: Aufschlagannahme mit unterschiedlichem Spin: Allerdings scheint die Intensität des Spiels nicht so intensiv zu sein wie Der alte Mann im Park. Für Roboter, Tischtennis
 Tokenisierung in einem Artikel verstehen!
Apr 12, 2024 pm 02:31 PM
Tokenisierung in einem Artikel verstehen!
Apr 12, 2024 pm 02:31 PM
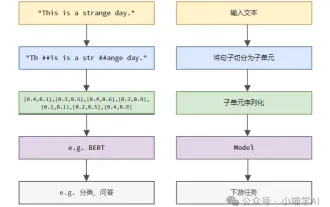
Sprachmodelle basieren auf Text, der normalerweise in Form von Zeichenfolgen vorliegt. Da die Eingabe in das Modell jedoch nur Zahlen sein kann, muss der Text in eine numerische Form umgewandelt werden. Die Tokenisierung ist eine grundlegende Aufgabe der Verarbeitung natürlicher Sprache. Sie kann eine fortlaufende Textsequenz (z. B. Sätze, Absätze usw.) entsprechend den spezifischen Anforderungen in eine Zeichenfolge (z. B. Wörter, Phrasen, Zeichen, Satzzeichen usw.) unterteilen. Die darin enthaltenen Einheiten werden als Token oder Wort bezeichnet. Gemäß dem in der Abbildung unten gezeigten spezifischen Prozess werden die Textsätze zunächst in Einheiten unterteilt, dann werden die einzelnen Elemente digitalisiert (in Vektoren abgebildet), dann werden diese Vektoren zur Codierung in das Modell eingegeben und schließlich an nachgelagerte Aufgaben ausgegeben erhalten Sie weiterhin das Endergebnis. Die Textsegmentierung kann entsprechend der Granularität der Textsegmentierung in Toke unterteilt werden.
 Um ein neues wissenschaftliches und komplexes Frage-Antwort-Benchmark- und Bewertungssystem für große Modelle bereitzustellen, haben UNSW, Argonne, die University of Chicago und andere Institutionen gemeinsam das SciQAG-Framework eingeführt
Jul 25, 2024 am 06:42 AM
Um ein neues wissenschaftliches und komplexes Frage-Antwort-Benchmark- und Bewertungssystem für große Modelle bereitzustellen, haben UNSW, Argonne, die University of Chicago und andere Institutionen gemeinsam das SciQAG-Framework eingeführt
Jul 25, 2024 am 06:42 AM
Herausgeber | Der Frage-Antwort-Datensatz (QA) von ScienceAI spielt eine entscheidende Rolle bei der Förderung der Forschung zur Verarbeitung natürlicher Sprache (NLP). Hochwertige QS-Datensätze können nicht nur zur Feinabstimmung von Modellen verwendet werden, sondern auch effektiv die Fähigkeiten großer Sprachmodelle (LLMs) bewerten, insbesondere die Fähigkeit, wissenschaftliche Erkenntnisse zu verstehen und zu begründen. Obwohl es derzeit viele wissenschaftliche QS-Datensätze aus den Bereichen Medizin, Chemie, Biologie und anderen Bereichen gibt, weisen diese Datensätze immer noch einige Mängel auf. Erstens ist das Datenformular relativ einfach, die meisten davon sind Multiple-Choice-Fragen. Sie sind leicht auszuwerten, schränken jedoch den Antwortauswahlbereich des Modells ein und können die Fähigkeit des Modells zur Beantwortung wissenschaftlicher Fragen nicht vollständig testen. Im Gegensatz dazu offene Fragen und Antworten
 Effiziente Parameter-Feinabstimmung umfangreicher Sprachmodelle – BitFit/Prefix/Prompt-Feinabstimmungsserie
Oct 07, 2023 pm 12:13 PM
Effiziente Parameter-Feinabstimmung umfangreicher Sprachmodelle – BitFit/Prefix/Prompt-Feinabstimmungsserie
Oct 07, 2023 pm 12:13 PM
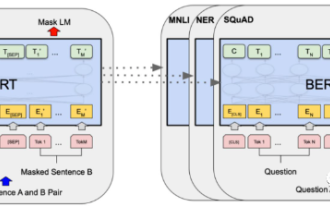
Im Jahr 2018 veröffentlichte Google BERT. Nach seiner Veröffentlichung übertraf es die State-of-the-Art-Ergebnisse (Sota) von 11 NLP-Aufgaben und stellte damit einen neuen Meilenstein in der NLP-Welt dar In der Abbildung unten ist der Trainingsprozess des BERT-Modells dargestellt, rechts der Feinabstimmungsprozess für bestimmte Aufgaben. Unter anderem dient die Feinabstimmungsphase der Feinabstimmung, wenn sie anschließend in einigen nachgelagerten Aufgaben verwendet wird, wie z. B. Textklassifizierung, Wortartkennzeichnung, Frage- und Antwortsysteme usw. BERT kann auf verschiedene Arten feinabgestimmt werden Aufgaben ohne Anpassung der Struktur. Durch das Aufgabendesign „vorab trainiertes Sprachmodell + Feinabstimmung der nachgelagerten Aufgabe“ werden leistungsstarke Modelleffekte erzielt. Seitdem ist „Sprachmodell vor dem Training + Feinabstimmung der nachgelagerten Aufgabe“ zum Mainstream-Training im NLP-Bereich geworden.
 Drei Geheimnisse für die Bereitstellung großer Modelle in der Cloud
Apr 24, 2024 pm 03:00 PM
Drei Geheimnisse für die Bereitstellung großer Modelle in der Cloud
Apr 24, 2024 pm 03:00 PM
Zusammenstellung|Produziert von Ich fange an, serverloses Cloud Computing zu vermissen. Ihre Anwendungen reichen von der Verbesserung der Konversations-KI bis hin zur Bereitstellung komplexer Analyselösungen für verschiedene Branchen und vielen anderen Funktionen. Viele Unternehmen setzen diese Modelle auf Cloud-Plattformen ein, da öffentliche Cloud-Anbieter bereits ein fertiges Ökosystem bereitstellen und dies der Weg des geringsten Widerstands ist. Allerdings ist es nicht billig. Die Cloud bietet darüber hinaus weitere Vorteile wie Skalierbarkeit, Effizienz und erweiterte Rechenfunktionen (GPUs auf Anfrage verfügbar). Es gibt einige wenig bekannte Aspekte der Bereitstellung von LLM auf öffentlichen Cloud-Plattformen
 AlphaFold 3 wird auf den Markt gebracht und sagt die Wechselwirkungen und Strukturen von Proteinen und allen Lebensmolekülen umfassend und mit weitaus größerer Genauigkeit als je zuvor voraus
Jul 16, 2024 am 12:08 AM
AlphaFold 3 wird auf den Markt gebracht und sagt die Wechselwirkungen und Strukturen von Proteinen und allen Lebensmolekülen umfassend und mit weitaus größerer Genauigkeit als je zuvor voraus
Jul 16, 2024 am 12:08 AM
Herausgeber | Rettichhaut Seit der Veröffentlichung des leistungsstarken AlphaFold2 im Jahr 2021 verwenden Wissenschaftler Modelle zur Proteinstrukturvorhersage, um verschiedene Proteinstrukturen innerhalb von Zellen zu kartieren, Medikamente zu entdecken und eine „kosmische Karte“ jeder bekannten Proteininteraktion zu zeichnen. Gerade hat Google DeepMind das AlphaFold3-Modell veröffentlicht, das gemeinsame Strukturvorhersagen für Komplexe wie Proteine, Nukleinsäuren, kleine Moleküle, Ionen und modifizierte Reste durchführen kann. Die Genauigkeit von AlphaFold3 wurde im Vergleich zu vielen dedizierten Tools in der Vergangenheit (Protein-Ligand-Interaktion, Protein-Nukleinsäure-Interaktion, Antikörper-Antigen-Vorhersage) deutlich verbessert. Dies zeigt, dass dies innerhalb eines einzigen einheitlichen Deep-Learning-Frameworks möglich ist
 RoSA: Eine neue Methode zur effizienten Feinabstimmung großer Modellparameter
Jan 18, 2024 pm 05:27 PM
RoSA: Eine neue Methode zur effizienten Feinabstimmung großer Modellparameter
Jan 18, 2024 pm 05:27 PM
Da Sprachmodelle in einem noch nie dagewesenen Ausmaß skaliert werden, wird eine umfassende Feinabstimmung für nachgelagerte Aufgaben unerschwinglich teuer. Um dieses Problem zu lösen, begannen Forscher, der PEFT-Methode Aufmerksamkeit zu schenken und sie zu übernehmen. Die Hauptidee der PEFT-Methode besteht darin, den Umfang der Feinabstimmung auf einen kleinen Satz von Parametern zu beschränken, um die Rechenkosten zu senken und gleichzeitig eine hochmoderne Leistung bei Aufgaben zum Verstehen natürlicher Sprache zu erzielen. Auf diese Weise können Forscher Rechenressourcen einsparen und gleichzeitig eine hohe Leistung aufrechterhalten, wodurch neue Forschungsschwerpunkte auf dem Gebiet der Verarbeitung natürlicher Sprache entstehen. RoSA ist eine neue PEFT-Technik, die durch Experimente mit einer Reihe von Benchmarks gezeigt hat, dass sie frühere Low-Rank-Adaptive- (LoRA) und reine Sparse-Feinabstimmungsmethoden mit demselben Parameterbudget übertrifft. Dieser Artikel wird näher darauf eingehen
 Den größten ViT der Geschichte bequem trainiert? Google aktualisiert das visuelle Sprachmodell PaLI: unterstützt mehr als 100 Sprachen
Apr 12, 2023 am 09:31 AM
Den größten ViT der Geschichte bequem trainiert? Google aktualisiert das visuelle Sprachmodell PaLI: unterstützt mehr als 100 Sprachen
Apr 12, 2023 am 09:31 AM
Der Fortschritt der Verarbeitung natürlicher Sprache ist in den letzten Jahren größtenteils auf groß angelegte Sprachmodelle zurückzuführen, die die Menge an Parametern und Trainingsdaten auf neue Höchstwerte bringen, und gleichzeitig werden die bestehenden Benchmark-Rankings geschlachtet! Beispielsweise veröffentlichte Google im April dieses Jahres das Sprachmodell PaLM (Pathways Language Model) mit 540 Milliarden Parametern, das Menschen in einer Reihe von Sprach- und Argumentationstests erfolgreich übertraf, insbesondere durch seine hervorragende Leistung in Lernszenarien mit wenigen Schüssen und kleinen Stichproben. PaLM gilt als Entwicklungsrichtung des Sprachmodells der nächsten Generation. Auf die gleiche Weise wirken visuelle Sprachmodelle tatsächlich Wunder, und die Leistung kann durch Erhöhen der Modellgröße verbessert werden. Natürlich, wenn es sich nur um ein multitaskingfähiges visuelles Sprachmodell handelt
