
 Backend-Entwicklung
Python-Tutorial
So installieren und verwenden Sie den genetischen Python-Algorithmus Geatpy
Backend-Entwicklung
Python-Tutorial
So installieren und verwenden Sie den genetischen Python-Algorithmus Geatpy
So installieren und verwenden Sie den genetischen Python-Algorithmus Geatpy

1. Installieren Sie geatpy zuerst mit dem Befehl pip3:
pip3 install geatpy
Die folgende Eingabeaufforderung wird angezeigt und die Installation ist erfolgreich:
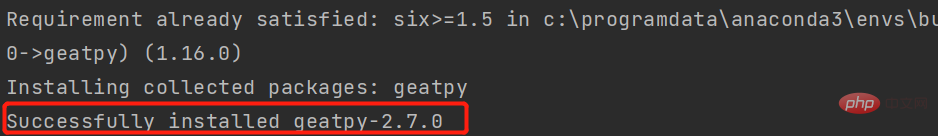 2. Die grundlegende Datenstruktur von geatpy
2. Die grundlegende Datenstruktur von geatpy
geatpy Die meisten Daten werden mithilfe von Numpy-Arrays gespeichert und berechnet. Im Folgenden werde ich vorstellen, wie die Konzepte in genetischen Algorithmen durch Numpy-Daten dargestellt werden und welche Bedeutung Zeilen und Spalten haben.
2.1 Populationschromosom
Das Wichtigste im genetischen Algorithmus ist die Chromosomendarstellung des Individuums. In Geatpy wird das Populationschromosom durch Chrom dargestellt, in dem jede Zeile dem Chromosom entspricht Der Code eines Individuums ist wie folgt aufgebaut: Dabei repräsentiert lind die Länge des Codes und Nind repräsentiert die Größe der Population (Anzahl der Individuen).
 2.2 Populationsphänotyp
2.2 Populationsphänotyp
Der Populationsphänotyp bezieht sich auf die Genphänotypmatrix Phen, die nach der Dekodierung der Populationschromosomenmatrix Chrom erhalten wird, und jede Spalte entspricht einer Entscheidungsvariablen Folgendes: Unter diesen stellt Nvar die Anzahl der Variablen dar. Der Wert von Phen hängt von der verwendeten Decodierungsmethode ab. Geatpy bietet eine Decodierungsmethode zum Konvertieren der Binär-/Gray-Code-Codierung in eine dezimale Ganzzahl oder eine reelle Zahl. Darüber hinaus kann Geatpy auch eine Population mit „Echtwertkodierung“ verwenden, die nicht dekodiert werden muss. Jedes Bit des Chromosoms dieser Population entspricht dem tatsächlichen Wert der Entscheidungsvariablen, dh Phen entspricht unter dieser Kodierung Chrom Verfahren.
2.3 Zielfunktionswert
Geatpy verwendet eine Numpy-Array-Typ-Matrix, um den Zielfunktionswert der Grundgesamtheit zu speichern. Jede Zeile wird allgemein als ObjV bezeichnet und entspricht jeder einzelnen Person. Daher entspricht jede Spalte einer Zielfunktion, sodass ObjV nur eine Spalte und bei Funktionen mit mehreren Zielen vorhanden ist , ObjV hat Für mehrere Spalten lautet die Darstellung von ObjV wie folgt: 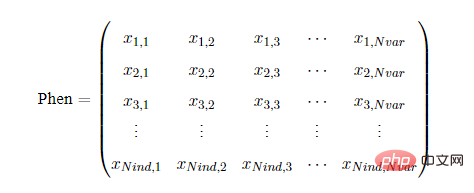
2.4 Individuelle Fitness
Geatpy verwendet Spaltenvektoren, um die individuelle Fitness der Bevölkerung zu speichern (berechnet durch die Fitnessfunktion). Im Allgemeinen FitnV genannt, ist es auch ein Array-Typ von Numpy, und jede Zeile entspricht jedem Individuum in der Populationsmatrix. Es hat also die gleiche Anzahl an Zeilen wie Chrom und das Format von FitnV ist wie folgt:
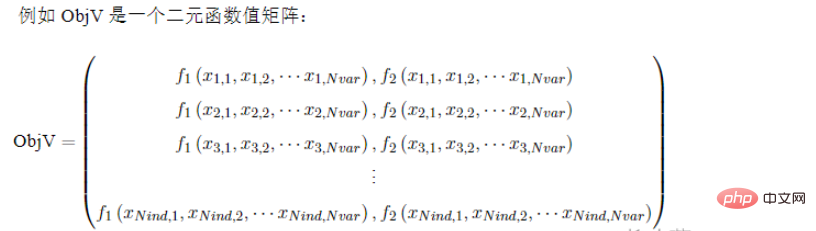
: Fitness in Geatpy folgt der Konvention „Minimale Fitness ist 0“.
2.5 Matrix für den Grad der Einschränkungsverletzung
Wenn ein Element der CV-Matrix kleiner oder gleich 0 ist, bedeutet dies, dass die Person dem Element entspricht erfüllt die entsprechende Einschränkung. Wenn er größer als 0 ist, bedeutet dies, dass die Einschränkungsbedingung verletzt wurde. Wenn der Wert größer als 0 ist, ist der Grad der Verletzung der Einschränkung durch die Person umso höher. Geatpy bietet zwei Methoden zum Umgang mit Einschränkungen: eine ist die Straffunktionsmethode und die andere ist die Machbarkeitsregel. Wenn Sie Machbarkeitsregeln verwenden, um mit Einschränkungen umzugehen, müssen Sie die CV-Matrix verwenden.
2.6 Dekodierungsmatrix
Die sogenannte Dekodierungsmatrix ist nur eine Matrix, die zur Beschreibung der Chromosomeneigenschaften der Population verwendet wird, z. B. des Bereichs der Entscheidungsvariablen, die durch jedes Element im Chromosom ausgedrückt werden, und ob es die Grenze enthält des Bereichs, ob es sich um einen Binär- oder Gray-Code handelt, ob eine logarithmische Skala verwendet werden soll, ob die durch das Chromosom nach der Dekodierung dargestellte Entscheidungsvariable eine kontinuierliche Variable oder eine diskrete Variable ist usw. 
- P: (Anordnungskodierung, d. h. die Elemente jedes Chromosoms unterscheiden sich voneinander)
注:’RI’和’P’编码的染色体都不需要解码,染色体上的每一位本身就代表着决策变量的真实值,因此“实整数编码”和“排列编码”可统称为“实值编码”
以BG编码为例,我们展示一下编译矩阵FieldD。FieldD的结构如下:
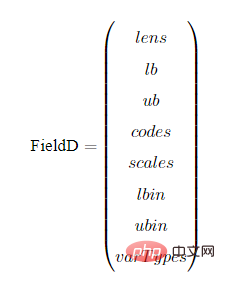
其中,lens,lb,ub,codes,scales,lbin,ubin,varTypes都是行向量,其长度等于决策变量的个数。
lens:代表以条染色体中,每个子染色体的长度。
lb:代表每个变量的上界
ub:代表每个变量的下界
codes:代表染色体字串用的编码方式,[1,0,1]代表第一个变量用的格雷编码,第二个变量用的二进制编码,第3个变量用的格雷编码。
scales:指明每个子串用的是算术刻度还是对数刻度。scales[i] = 0为算术刻度,scales[i] = 1为对数刻度(对数刻度很少用,可以忽略。)
lbin:代表变量上界是否包含其范围边界。0代表不包含,1代表包含。‘[ ’和 ‘(’ 的区别
ubin:代表变量下界是否包含其范围边界。0代表不包含,1代表包含。
varTypes:代表决策变量的类型,元素为0表示对应位置的决策变量是连续型变量;1表示对应的是离散型变量。
例如:有以下一个译码矩阵

它表示待解码的种群染色体矩阵Chrom解码后可以表示成3个决策变量,每个决策变量的取值范围分别是[1,10], [2,9], [3,15]。其中第一第二个变量采用的是二进制编码,第三个变量采用的是格雷编码,且第一、第三个决策变量为连续型变量;第二个为离散型变量。
#通过种群染色体chrom和译码矩阵FieldD,可解码成种群表现型矩阵。 import geatpy as ea Phen = ea.bs2ri(Chrom, FieldD)
2.7 进化追踪器
在使用Geatpy进行进化算法编程时,常常建立一个进化追踪器(如pop_trace)来记录种群在进化的过程中各代的最优个体,尤其是采用无精英保留机制时,进化追踪器帮助我们记录种群在进化过程中的最优个体。待进化完成后,再从进化追踪器中挑选出“历史最优”的个体。这种进化记录器有多种,其中一种是numpy的array类型的,结构如下:其中MAXGEN是种群进化的代数(迭代次数)。
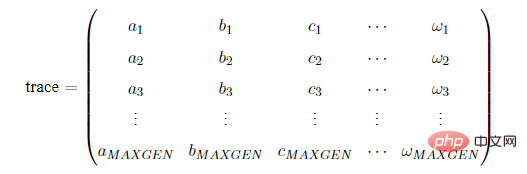
trace的每一列代表不同的指标,比如第一列记录各代种群的最佳目标函数值,第二列记录各代种群的平均目标函数值…trace的每一行对应每一代,如第一行代表第一代,第二行代表第二代…另外一种进化记录器是一个列表,列表中的每一个元素都是一个拥有相同数据类型的数据。比如在Geatpy的面向对象进化算法框架中的pop_trace,它是一个列表,列表中的每一个元素都是历代的种群对象。
3. geatpy的种群结构
3.1 Population类
在Geatpy提供的面向对象进化算法框架中,种群类(Population)是一个存储着与种群个体相关信息的类。它有以下基本属性:
sizes : int -种群规模,即种群的个体数目。
ChromNum : int -染色体的数目,即每个个体有多少条染色体。
Encoding : str -染色体编码方式。
Field : array -译码矩阵,可以是FieldD或FieldDR。
Chrom : array -种群染色体矩阵,每一行对应一个个体的一条染色体。
Lind : int -种群染色体长度。
ObjV : array -种群目标函数值矩阵。
FitnV : array -种群个体适应度列向量。
CV : array -种群个体违反约束条件程度的矩阵。
Phen : array -种群表现型矩阵。
可以直接对种群对象进行提取个体、个体合并等操作,比如pop1和pop2是两个种群对象,则通过语句“pop3 = pop1 + pop2”,即可把两个种群的个体合并,得到一个新的种群。在合并的过程中,实际上是把种群的各个属性进行合并,然后用合并的数据来生成一个新的种群(详见Population.py)。又比如执行语句“pop3 = pop1[[0]]”,可以把种群的第0号个体抽取出来,得到一个新的只有一个个体的种群对象pop3。值得注意的是,种群的这种个体抽取操作要求下标必须为列表或是Numpy array类型的行向量,不能是标量(详见Population.py)
3.2 PsyPopulation类
PsyPopulation类是Population的子类,它提供Population类所不支持的多染色体混合编码。它有以下基本属性:
sizes : int -种群规模,即种群的个体数目。
ChromNum : int -染色体的数目,即每个个体有多少条染色体。
Encodings : list -存储各染色体编码方式的列表。
Fields : list -存储各染色体对应的译码矩阵的列表。
Chroms : list -存储种群各染色体矩阵的列表。
Linds : list -存储种群各染色体长度的列表。
ObjV : array -种群目标函数值矩阵。
FitnV : array -种群个体适应度列向量。
CV : array -种群个体违反约束条件程度的矩阵。
Phen : array -种群表现型矩阵。
可见PsyPopulation类基本与Population类一样,不同之处是采用Linds、Encodings、Fields和Chroms分别存储多个Lind、Encoding、Field和Chrom。
PsyPopulation类的对象往往与带“psy”字样的进化算法模板配合使用,以实现多染色体混合编码的进化优化。
4. 求解标准测试函数——McCormick函数
遗传算法求解以下函数的最小值:
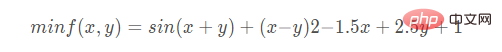
代码实现:
#-*-coding:utf-8-*-
import numpy as np
import geatpy as ea#导入geatpy库
import time
"""============================目标函数============================"""
def aim(Phen):#传入种群染色体矩阵解码后的基因表现型矩阵
x1 = Phen[:, [0]]#取出第一列,得到所有个体的第一个自变量
x2 = Phen[:, [1]]#取出第二列,得到所有个体的第二个自变量
return np.sin(x1 + x2) + (x1 - x2) ** 2 - 1.5 * x1 + 2.5 * x2+1
"""============================变量设置============================"""
x1 = [-1.5, 4]#第一个决策变量范围
x2 = [-3, 4]#第二个决策变量范围
b1 = [1, 1]#第一个决策变量边界,1表示包含范围的边界,0表示不包含
b2 = [1, 1]#第二个决策变量边界,1表示包含范围的边界,0表示不包含
#生成自变量的范围矩阵,使得第一行为所有决策变量的下界,第二行为上界
ranges=np.vstack([x1, x2]).T
#生成自变量的边界矩阵
borders=np.vstack([b1, b2]).T
varTypes = np.array([0, 0])#决策变量的类型,0表示连续,1表示离散
"""==========================染色体编码设置========================="""
Encoding ='BG'#'BG'表示采用二进制/格雷编码
codes = [1, 1]#决策变量的编码方式,两个1表示变量均使用格雷编码
precisions =[6, 6]#决策变量的编码精度,表示解码后能表示的决策变量的精度可达到小数点后6位
scales = [0, 0]#0表示采用算术刻度,1表示采用对数刻度#调用函数创建译码矩阵
FieldD =ea.crtfld(Encoding,varTypes,ranges,borders,precisions,codes,scales)
"""=========================遗传算法参数设置========================"""
NIND = 20#种群个体数目
MAXGEN = 100#最大遗传代数
maxormins = np.array([1])#表示目标函数是最小化,元素为-1则表示对应的目标函数是最大化
selectStyle ='sus'#采用随机抽样选择
recStyle ='xovdp'#采用两点交叉
mutStyle ='mutbin'#采用二进制染色体的变异算子
Lind =int(np.sum(FieldD[0, :]))#计算染色体长度
pc= 0.9#交叉概率
pm= 1/Lind#变异概率
obj_trace = np.zeros((MAXGEN, 2))#定义目标函数值记录器
var_trace = np.zeros((MAXGEN, Lind))#染色体记录器,记录历代最优个体的染色体
"""=========================开始遗传算法进化========================"""
start_time = time.time()#开始计时
Chrom = ea.crtpc(Encoding,NIND, FieldD)#生成种群染色体矩阵
variable = ea.bs2ri(Chrom, FieldD)#对初始种群进行解码
ObjV = aim(variable)#计算初始种群个体的目标函数值
best_ind = np.argmin(ObjV)#计算当代最优个体的序号
#开始进化
for gen in range(MAXGEN):
FitnV = ea.ranking(maxormins * ObjV)#根据目标函数大小分配适应度值
SelCh = Chrom[ea.selecting(selectStyle,FitnV,NIND-1),:]#选择
SelCh = ea.recombin(recStyle, SelCh, pc)#重组
SelCh = ea.mutate(mutStyle, Encoding, SelCh, pm)#变异
# #把父代精英个体与子代的染色体进行合并,得到新一代种群
Chrom = np.vstack([Chrom[best_ind, :], SelCh])
Phen = ea.bs2ri(Chrom, FieldD)#对种群进行解码(二进制转十进制)
ObjV = aim(Phen)#求种群个体的目标函数值
#记录
best_ind = np.argmin(ObjV)#计算当代最优个体的序号
obj_trace[gen,0]=np.sum(ObjV)/ObjV.shape[0]#记录当代种群的目标函数均值
obj_trace[gen,1]=ObjV[best_ind]#记录当代种群最优个体目标函数值
var_trace[gen,:]=Chrom[best_ind,:]#记录当代种群最优个体的染色体
# 进化完成
end_time = time.time()#结束计时
ea.trcplot(obj_trace, [['种群个体平均目标函数值','种群最优个体目标函数值']])#绘制图像
"""============================输出结果============================"""
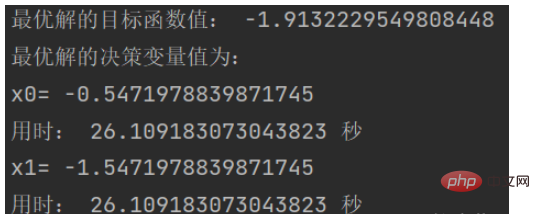
best_gen = np.argmin(obj_trace[:, [1]])
print('最优解的目标函数值:', obj_trace[best_gen, 1])
variable = ea.bs2ri(var_trace[[best_gen], :], FieldD)#解码得到表现型(即对应的决策变量值)
print('最优解的决策变量值为:')
for i in range(variable.shape[1]):
print('x'+str(i)+'=',variable[0, i])
print('用时:', end_time - start_time,'秒')效果图:
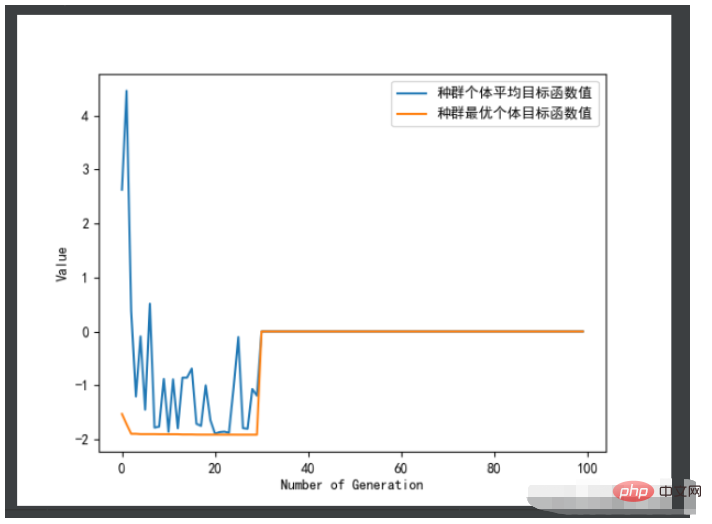
结果如下:
Das obige ist der detaillierte Inhalt vonSo installieren und verwenden Sie den genetischen Python-Algorithmus Geatpy. Für weitere Informationen folgen Sie bitte anderen verwandten Artikeln auf der PHP chinesischen Website!

Heiße KI -Werkzeuge

Undresser.AI Undress
KI-gestützte App zum Erstellen realistischer Aktfotos

AI Clothes Remover
Online-KI-Tool zum Entfernen von Kleidung aus Fotos.

Undress AI Tool
Ausziehbilder kostenlos

Clothoff.io
KI-Kleiderentferner

Video Face Swap
Tauschen Sie Gesichter in jedem Video mühelos mit unserem völlig kostenlosen KI-Gesichtstausch-Tool aus!

Heißer Artikel
Heiße Werkzeuge

Notepad++7.3.1
Einfach zu bedienender und kostenloser Code-Editor

SublimeText3 chinesische Version
Chinesische Version, sehr einfach zu bedienen

Senden Sie Studio 13.0.1
Leistungsstarke integrierte PHP-Entwicklungsumgebung

Dreamweaver CS6
Visuelle Webentwicklungstools

SublimeText3 Mac-Version
Codebearbeitungssoftware auf Gottesniveau (SublimeText3)
Heiße Themen



 PHP und Python: Verschiedene Paradigmen erklärt
Apr 18, 2025 am 12:26 AM
PHP und Python: Verschiedene Paradigmen erklärt
Apr 18, 2025 am 12:26 AM
PHP ist hauptsächlich prozedurale Programmierung, unterstützt aber auch die objektorientierte Programmierung (OOP). Python unterstützt eine Vielzahl von Paradigmen, einschließlich OOP, funktionaler und prozeduraler Programmierung. PHP ist für die Webentwicklung geeignet, und Python eignet sich für eine Vielzahl von Anwendungen wie Datenanalyse und maschinelles Lernen.
 Wählen Sie zwischen PHP und Python: Ein Leitfaden
Apr 18, 2025 am 12:24 AM
Wählen Sie zwischen PHP und Python: Ein Leitfaden
Apr 18, 2025 am 12:24 AM
PHP eignet sich für Webentwicklung und schnelles Prototyping, und Python eignet sich für Datenwissenschaft und maschinelles Lernen. 1.PHP wird für die dynamische Webentwicklung verwendet, mit einfacher Syntax und für schnelle Entwicklung geeignet. 2. Python hat eine kurze Syntax, ist für mehrere Felder geeignet und ein starkes Bibliotheksökosystem.
 Python vs. JavaScript: Die Lernkurve und Benutzerfreundlichkeit
Apr 16, 2025 am 12:12 AM
Python vs. JavaScript: Die Lernkurve und Benutzerfreundlichkeit
Apr 16, 2025 am 12:12 AM
Python eignet sich besser für Anfänger mit einer reibungslosen Lernkurve und einer kurzen Syntax. JavaScript ist für die Front-End-Entwicklung mit einer steilen Lernkurve und einer flexiblen Syntax geeignet. 1. Python-Syntax ist intuitiv und für die Entwicklung von Datenwissenschaften und Back-End-Entwicklung geeignet. 2. JavaScript ist flexibel und in Front-End- und serverseitiger Programmierung weit verbreitet.
 Kann gegen Code in Windows 8 ausgeführt werden
Apr 15, 2025 pm 07:24 PM
Kann gegen Code in Windows 8 ausgeführt werden
Apr 15, 2025 pm 07:24 PM
VS -Code kann unter Windows 8 ausgeführt werden, aber die Erfahrung ist möglicherweise nicht großartig. Stellen Sie zunächst sicher, dass das System auf den neuesten Patch aktualisiert wurde, und laden Sie dann das VS -Code -Installationspaket herunter, das der Systemarchitektur entspricht und sie wie aufgefordert installiert. Beachten Sie nach der Installation, dass einige Erweiterungen möglicherweise mit Windows 8 nicht kompatibel sind und nach alternativen Erweiterungen suchen oder neuere Windows -Systeme in einer virtuellen Maschine verwenden müssen. Installieren Sie die erforderlichen Erweiterungen, um zu überprüfen, ob sie ordnungsgemäß funktionieren. Obwohl VS -Code unter Windows 8 möglich ist, wird empfohlen, auf ein neueres Windows -System zu upgraden, um eine bessere Entwicklungserfahrung und Sicherheit zu erzielen.
 PHP und Python: Ein tiefes Eintauchen in ihre Geschichte
Apr 18, 2025 am 12:25 AM
PHP und Python: Ein tiefes Eintauchen in ihre Geschichte
Apr 18, 2025 am 12:25 AM
PHP entstand 1994 und wurde von Rasmuslerdorf entwickelt. Es wurde ursprünglich verwendet, um Website-Besucher zu verfolgen und sich nach und nach zu einer serverseitigen Skriptsprache entwickelt und in der Webentwicklung häufig verwendet. Python wurde Ende der 1980er Jahre von Guidovan Rossum entwickelt und erstmals 1991 veröffentlicht. Es betont die Lesbarkeit und Einfachheit der Code und ist für wissenschaftliche Computer, Datenanalysen und andere Bereiche geeignet.
 So führen Sie Programme in der terminalen VSCODE aus
Apr 15, 2025 pm 06:42 PM
So führen Sie Programme in der terminalen VSCODE aus
Apr 15, 2025 pm 06:42 PM
Im VS -Code können Sie das Programm im Terminal in den folgenden Schritten ausführen: Erstellen Sie den Code und öffnen Sie das integrierte Terminal, um sicherzustellen, dass das Codeverzeichnis mit dem Terminal Working -Verzeichnis übereinstimmt. Wählen Sie den Befehl aus, den Befehl ausführen, gemäß der Programmiersprache (z. B. Pythons Python your_file_name.py), um zu überprüfen, ob er erfolgreich ausgeführt wird, und Fehler auflösen. Verwenden Sie den Debugger, um die Debugging -Effizienz zu verbessern.
 Ist die VSCODE -Erweiterung bösartig?
Apr 15, 2025 pm 07:57 PM
Ist die VSCODE -Erweiterung bösartig?
Apr 15, 2025 pm 07:57 PM
VS -Code -Erweiterungen stellen böswillige Risiken dar, wie das Verstecken von böswilligem Code, das Ausbeutetieren von Schwachstellen und das Masturbieren als legitime Erweiterungen. Zu den Methoden zur Identifizierung böswilliger Erweiterungen gehören: Überprüfung von Verlegern, Lesen von Kommentaren, Überprüfung von Code und Installation mit Vorsicht. Zu den Sicherheitsmaßnahmen gehören auch: Sicherheitsbewusstsein, gute Gewohnheiten, regelmäßige Updates und Antivirensoftware.
 Kann Visual Studio -Code in Python verwendet werden
Apr 15, 2025 pm 08:18 PM
Kann Visual Studio -Code in Python verwendet werden
Apr 15, 2025 pm 08:18 PM
VS -Code kann zum Schreiben von Python verwendet werden und bietet viele Funktionen, die es zu einem idealen Werkzeug für die Entwicklung von Python -Anwendungen machen. Sie ermöglichen es Benutzern: Installation von Python -Erweiterungen, um Funktionen wie Code -Abschluss, Syntax -Hervorhebung und Debugging zu erhalten. Verwenden Sie den Debugger, um Code Schritt für Schritt zu verfolgen, Fehler zu finden und zu beheben. Integrieren Sie Git für die Versionskontrolle. Verwenden Sie Tools für die Codeformatierung, um die Codekonsistenz aufrechtzuerhalten. Verwenden Sie das Lining -Tool, um potenzielle Probleme im Voraus zu erkennen.
