
 Technologie-Peripheriegeräte
KI
Finden Sie die „anfälligsten Individuen' aus den „seltensten Mutationen', und KI sagt die Pathogenität genetischer Varianten genau voraus
Technologie-Peripheriegeräte
KI
Finden Sie die „anfälligsten Individuen' aus den „seltensten Mutationen', und KI sagt die Pathogenität genetischer Varianten genau voraus
Finden Sie die „anfälligsten Individuen' aus den „seltensten Mutationen', und KI sagt die Pathogenität genetischer Varianten genau voraus

Normalerweise trägt fast jeder Dutzende potenziell schädlicher seltener Varianten in sich. Eine der größten Schwierigkeiten bei der klinischen Beurteilung häufiger Varianten ist die Unfähigkeit, Personen effektiv zu identifizieren, bei denen ein hohes Risiko für die Entwicklung der Krankheit besteht. Bei der Forschung des Primate Genome Project verwendeten Wissenschaftler das neuronale Netzwerk PrimateAI-3D mit künstlicher Intelligenz, um hochpathogene seltene Mutationen durch Evolutionsanalyse zu lokalisieren, mit der Idee, „die seltensten Mutationen zu verwenden, um die Individuen zu finden, die am anfälligsten für Krankheiten sind“. Wird verwendet, um das Krankheitsrisiko einer Person vorherzusagen.
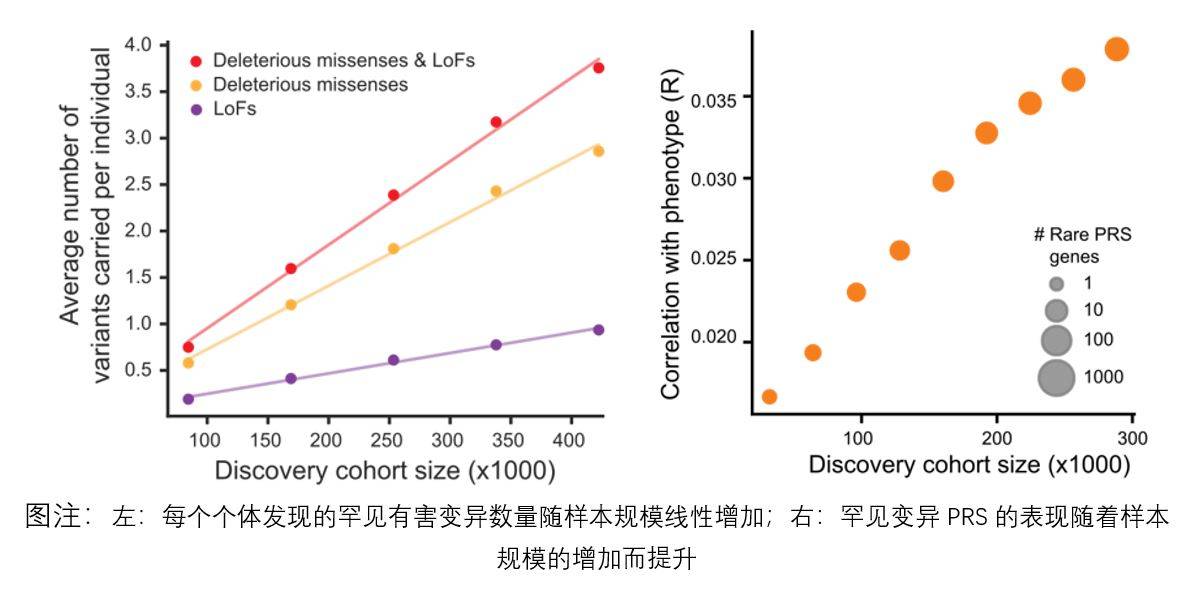
Das Team von Professor Tomas Marquez-Bonet aus Spanien und dem Illumina Artificial Intelligence Laboratory führte gemeinsam mehrere Forschungsgruppen durch und verglich die gesamten Genomsequenzierungsdaten von insgesamt 809 Proben von 233 Primatenarten, um Menschen zu identifizieren. Es gibt 4,3 Millionen genetische Mutationen Stellen auf orthologen Proteinen, die zu Veränderungen in der Proteinstruktur führen können.
Die Forscher verwendeten die oben genannten genetischen Mutationsstellen als Grundlage des Datensatzes, fügten sie den genetischen Daten menschlicher Krankheiten hinzu und trainierten das neuronale Netzwerk der künstlichen Intelligenz PrimateAI-3D mit einem Gendatensatz, der 4,5 Millionen Arten möglicher gutartiger Mutationen enthielt um die Pathogenität genetischer Varianten genauer vorhersagen zu können.
Genvariation ist eine der Hauptursachen für Krankheiten. Basierend auf der genetischen Verwandtschaft zwischen nichtmenschlichen Primaten und Menschen können dieselben genetischen Mutationen zu ähnlichen Ergebnissen führen, und Mutationen, die bei Primaten häufig vorkommen, können bedeuten, dass diese Mutationen eher harmlos sind oder nur einen äußerst begrenzten Schaden anrichten.
Wie kann man also das Risiko häufiger Krankheiten wie Diabetes und Herz-Kreislauf-Erkrankungen vorhersagen, die durch genetische Faktoren einer Person verursacht werden? Ist es besser, als Summe von Tausenden häufiger genetischer Varianten zu werten, die einen kleinen Effekt haben, oder als Summe einiger weniger seltener Mutationen, die einen signifikanten Effekt haben?
Umfassende Untersuchungen zeigen, dass häufige und seltene Varianten eine komplementäre Rolle bei der Vorhersage des Krankheitsrisikos beim Menschen spielen. Häufige Mutationen identifizieren mehr Personen, die wahrscheinlich an der Krankheit leiden, während seltene Mutationen es einfacher machen, die abnormalen Personen mit dem höchsten Risiko zu finden. Daher ist die Einbeziehung seltener Varianten in die klinische Beurteilung möglicherweise besser als die Verwendung nur häufiger Varianten, um die extremen Personen zu identifizieren, die letztendlich von den meisten Krankheiten betroffen sind und die am meisten einer Behandlung bedürfen oder an einer schweren Erkrankung im Frühstadium leiden zur Vorsorgeuntersuchung.
Diese Studie demonstriert erfolgreich die Anwendung der Kombination von Sequenzierungsdaten von Primaten mit Deep-Learning-Modellen, die uns helfen, die Pathogenität menschlicher genetischer Variationen zu verstehen und dazu beitragen können, dass die personalisierte Genommedizin eine bessere diagnostische Anleitung im klinischen Umfeld bietet.
Autor: Gemeinsam erstellt und zusammengestellt von der Forschungsgruppe von Professor Zhang Guojie von der Zhejiang-Universität
Herausgeber: Xu Qimin
*Exklusives Manuskript von Wenhui, bitte geben Sie beim Nachdruck die Quelle an.
Das obige ist der detaillierte Inhalt vonFinden Sie die „anfälligsten Individuen' aus den „seltensten Mutationen', und KI sagt die Pathogenität genetischer Varianten genau voraus. Für weitere Informationen folgen Sie bitte anderen verwandten Artikeln auf der PHP chinesischen Website!

Heiße KI -Werkzeuge

Undresser.AI Undress
KI-gestützte App zum Erstellen realistischer Aktfotos

AI Clothes Remover
Online-KI-Tool zum Entfernen von Kleidung aus Fotos.

Undress AI Tool
Ausziehbilder kostenlos

Clothoff.io
KI-Kleiderentferner

AI Hentai Generator
Erstellen Sie kostenlos Ai Hentai.

Heißer Artikel
Heiße Werkzeuge

Notepad++7.3.1
Einfach zu bedienender und kostenloser Code-Editor

SublimeText3 chinesische Version
Chinesische Version, sehr einfach zu bedienen

Senden Sie Studio 13.0.1
Leistungsstarke integrierte PHP-Entwicklungsumgebung

Dreamweaver CS6
Visuelle Webentwicklungstools

SublimeText3 Mac-Version
Codebearbeitungssoftware auf Gottesniveau (SublimeText3)
Heiße Themen
 1376
1376
 52
52

 Ich habe versucht, die Vibe -Codierung mit Cursor AI und es ist erstaunlich!
Mar 20, 2025 pm 03:34 PM
Ich habe versucht, die Vibe -Codierung mit Cursor AI und es ist erstaunlich!
Mar 20, 2025 pm 03:34 PM
Die Vibe -Codierung verändert die Welt der Softwareentwicklung, indem wir Anwendungen mit natürlicher Sprache anstelle von endlosen Codezeilen erstellen können. Inspiriert von Visionären wie Andrej Karpathy, lässt dieser innovative Ansatz Dev
 Top 5 Genai Starts vom Februar 2025: GPT-4,5, GROK-3 & MEHR!
Mar 22, 2025 am 10:58 AM
Top 5 Genai Starts vom Februar 2025: GPT-4,5, GROK-3 & MEHR!
Mar 22, 2025 am 10:58 AM
Februar 2025 war ein weiterer bahnbrechender Monat für die Generative KI, die uns einige der am meisten erwarteten Modell-Upgrades und bahnbrechenden neuen Funktionen gebracht hat. Von Xais Grok 3 und Anthropics Claude 3.7 -Sonett, um g zu eröffnen
 Wie benutze ich Yolo V12 zur Objekterkennung?
Mar 22, 2025 am 11:07 AM
Wie benutze ich Yolo V12 zur Objekterkennung?
Mar 22, 2025 am 11:07 AM
Yolo (Sie schauen nur einmal) war ein führender Echtzeit-Objekterkennungsrahmen, wobei jede Iteration die vorherigen Versionen verbessert. Die neueste Version Yolo V12 führt Fortschritte vor, die die Genauigkeit erheblich verbessern
 Ist Chatgpt 4 o verfügbar?
Mar 28, 2025 pm 05:29 PM
Ist Chatgpt 4 o verfügbar?
Mar 28, 2025 pm 05:29 PM
Chatgpt 4 ist derzeit verfügbar und weit verbreitet, wodurch im Vergleich zu seinen Vorgängern wie ChatGPT 3.5 signifikante Verbesserungen beim Verständnis des Kontextes und des Generierens kohärenter Antworten zeigt. Zukünftige Entwicklungen können mehr personalisierte Inters umfassen
 Gencast von Google: Wettervorhersage mit Gencast Mini Demo
Mar 16, 2025 pm 01:46 PM
Gencast von Google: Wettervorhersage mit Gencast Mini Demo
Mar 16, 2025 pm 01:46 PM
Gencast von Google Deepmind: Eine revolutionäre KI für die Wettervorhersage Die Wettervorhersage wurde einer dramatischen Transformation unterzogen, die sich von rudimentären Beobachtungen zu ausgefeilten AI-angetriebenen Vorhersagen überschreitet. Google DeepMinds Gencast, ein Bodenbrei
 Welche KI ist besser als Chatgpt?
Mar 18, 2025 pm 06:05 PM
Welche KI ist besser als Chatgpt?
Mar 18, 2025 pm 06:05 PM
Der Artikel erörtert KI -Modelle, die Chatgpt wie Lamda, Lama und Grok übertreffen und ihre Vorteile in Bezug auf Genauigkeit, Verständnis und Branchenauswirkungen hervorheben. (159 Charaktere)
 O1 gegen GPT-4O: Ist OpenAIs neues Modell besser als GPT-4O?
Mar 16, 2025 am 11:47 AM
O1 gegen GPT-4O: Ist OpenAIs neues Modell besser als GPT-4O?
Mar 16, 2025 am 11:47 AM
Openais O1: Ein 12-tägiger Geschenkbummel beginnt mit ihrem bisher mächtigsten Modell Die Ankunft im Dezember bringt eine globale Verlangsamung, Schneeflocken in einigen Teilen der Welt, aber Openai fängt gerade erst an. Sam Altman und sein Team starten ein 12-tägiges Geschenk Ex
 Beste KI -Kunstgeneratoren (kostenlos & amp; bezahlt) für kreative Projekte
Apr 02, 2025 pm 06:10 PM
Beste KI -Kunstgeneratoren (kostenlos & amp; bezahlt) für kreative Projekte
Apr 02, 2025 pm 06:10 PM
Der Artikel überprüft Top -KI -Kunstgeneratoren, diskutiert ihre Funktionen, Eignung für kreative Projekte und Wert. Es zeigt MidJourney als den besten Wert für Fachkräfte und empfiehlt Dall-E 2 für hochwertige, anpassbare Kunst.
