
 Technologie-Peripheriegeräte
KI
Weltneuheit: Der neue Open-Source-KI-Algorithmus von Molecular Heart zur Überwindung der Probleme der Proteinseitenkettenvorhersage und des Sequenzdesigns
Technologie-Peripheriegeräte
KI
Weltneuheit: Der neue Open-Source-KI-Algorithmus von Molecular Heart zur Überwindung der Probleme der Proteinseitenkettenvorhersage und des Sequenzdesigns
Weltneuheit: Der neue Open-Source-KI-Algorithmus von Molecular Heart zur Überwindung der Probleme der Proteinseitenkettenvorhersage und des Sequenzdesigns

Die Bildung der Proteinstruktur und -funktion hängt weitgehend von der Wechselwirkung zwischen Seitenkettenatomen ab. Daher ist die genaue Proteinseitenkettenvorhersage (PSCP) ein Schlüsselelement bei der Lösung der Probleme der Proteinstrukturvorhersage und des Proteindesigns. Frühere Proteinstrukturvorhersagen konzentrierten sich jedoch hauptsächlich auf die Hauptkettenstruktur, und die Vorhersage der Seitenkettenstruktur war schon immer ein schwieriges Problem, das nicht vollständig gelöst wurde.
Kürzlich hat das Team von Molecular Heart Ein KI-Algorithmus, der gleichzeitig eine Proteinseitenkettenvorhersage und ein Sequenzdesign durchführen kann.
Das Papier wurde in den Proceedings of the National Academy of Sciences (PNAS) veröffentlicht und das vorab trainierte Modell, der Quellcode und die Inferenzskripte wurden als Open Source auf Github bereitgestellt. ?? Quelllink:https:/ / github.com/MattMcPartlon/AttnPacker
Hintergrund
- Die meisten aktuellen Algorithmen zur Vorhersage der Proteinstruktur konzentrieren sich hauptsächlich auf die Strukturanalyse der Hauptkette, aber die Vorhersage der Proteinseitenkettenstruktur ist immer noch ein Problem, das nicht vollständig gelöst wurde. Unabhängig davon, ob es sich um beliebte Algorithmen zur Vorhersage der Proteinstruktur wie AlphaFold2 oder um Algorithmen mit Fokus auf die Vorhersage der Seitenkettenstruktur wie DLPacker und RosettaPacker handelt, sind weder die Genauigkeit noch die Geschwindigkeit zufriedenstellend. Dies bringt auch Einschränkungen für das Proteindesign mit sich. Traditionelle Methoden wie RosettaPacker verwenden hauptsächlich Methoden zur Energieoptimierung, bei denen zunächst die Verteilung der Seitenkettenatome gruppiert wird und dann die Gruppierung der Seitenketten nach einer bestimmten Aminosäure durchsucht wird, um die Kombination mit der kleinsten Energie zu finden. Diese Methoden unterscheiden sich in erster Linie von der Auswahl der Rotamer-Bibliotheken, Energiefunktionen und Energieminimierungsverfahren durch den Forscher, wobei die Genauigkeit durch die Verwendung von Suchheuristiken und diskreten Stichprobenverfahren begrenzt ist. In der Branche gibt es auch Side-Chain-Vorhersagemethoden, die auf Deep Learning basieren, beispielsweise DLPacker, das PSCP als Bild-zu-Bild-Konvertierungsproblem formuliert und eine U-Net-Modellstruktur übernimmt. Allerdings sind die Vorhersagegenauigkeit und -geschwindigkeit noch nicht optimal.
- Methode AttnPacker ist eine End-to-End-Deep-Learning-Methode zur Vorhersage von Proteinseitenkettenkoordinaten. Es simuliert gemeinsam Seitenkettenwechselwirkungen mit direkt vorhergesagten Seitenkettenstrukturen, die physikalisch besser realisierbar sind, mit weniger Atomkollisionen und idealeren Bindungslängen und -winkeln.
Konkret führt AttnPacker eine Tiefenkartenkonverterarchitektur ein, die die geometrischen und relationalen Aspekte von PSCP nutzt. Inspiriert von AlphaFold2 schlägt Molecular Heart positionsbewusste Dreiecksaktualisierungen vor, um paarweise Merkmale mithilfe eines graphbasierten Frameworks zur Berechnung der Dreiecksaufmerksamkeit und multiplikativer Aktualisierungen zu optimieren. Mit diesem Ansatz verfügt AttnPacker über deutlich weniger Speicher und ein Modell mit höherer Kapazität. Darüber hinaus untersucht Molecular Heart mehrere SE (3) äquivariante Aufmerksamkeitsmechanismen und schlägt eine äquivariante Transformatorarchitektur für das Lernen aus 3D-Punkten vor.
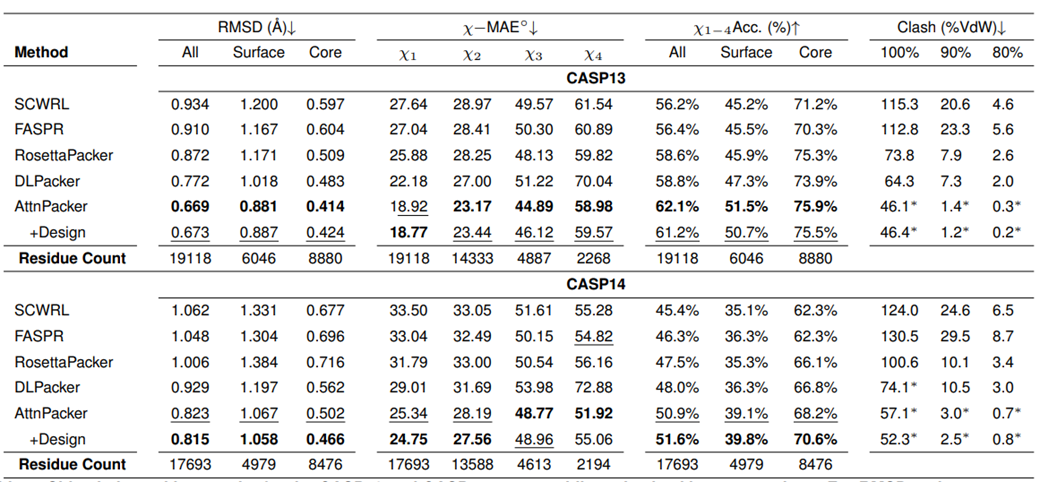
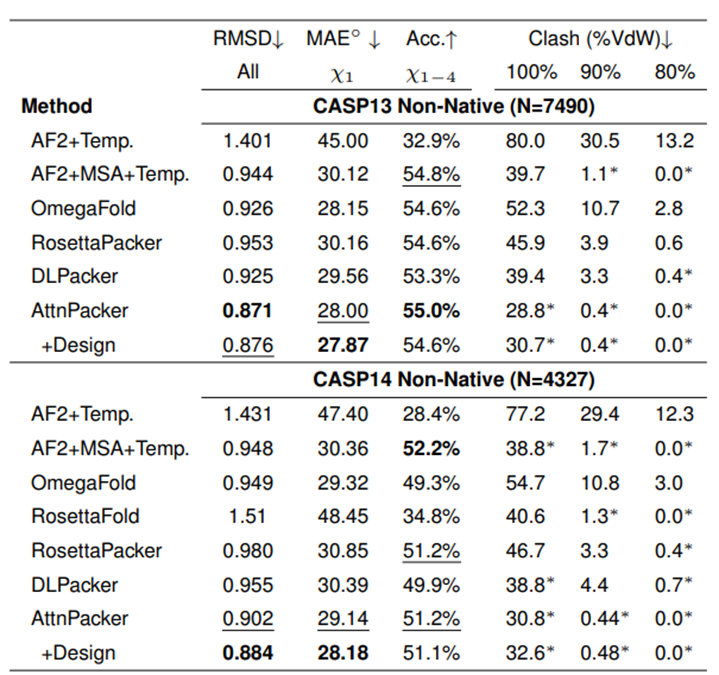
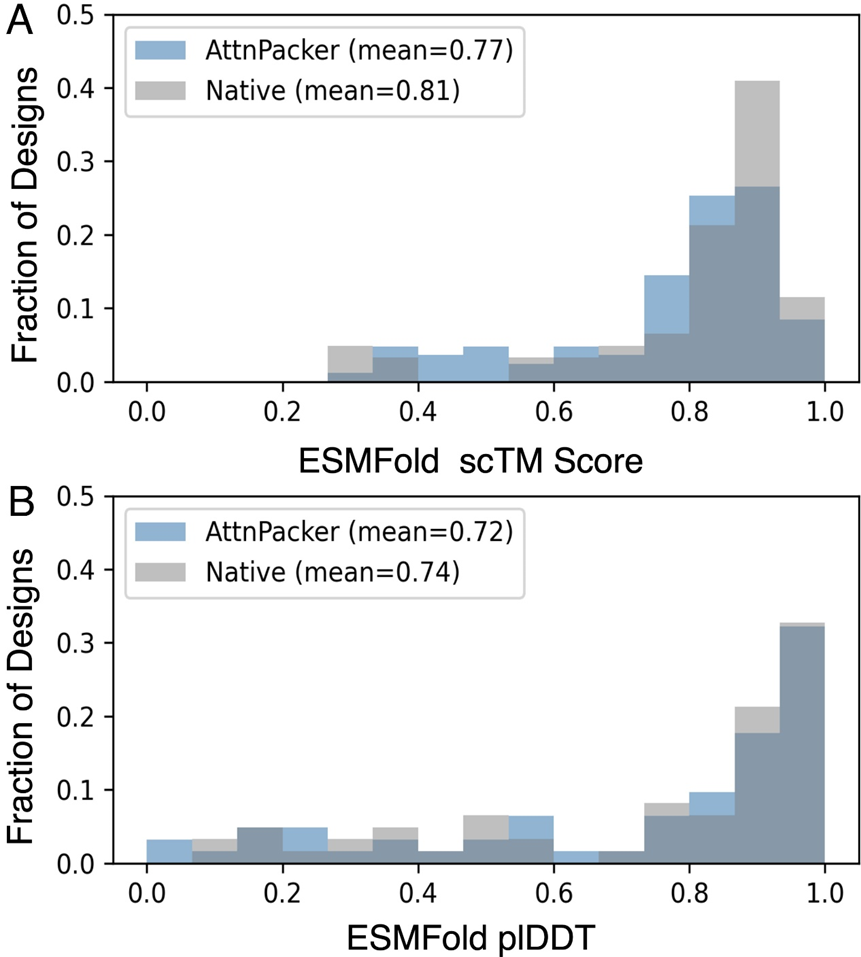
AttnPacker führt den Prozess aus. Die Koordinaten und die Sequenz des Proteinrückgrats werden als Eingabe verwendet, und die räumliche Merkmalskarte und die Äquivariablenbasis werden basierend auf den Koordinateninformationen abgeleitet. Die Feature-Map wird vom invarianten Graph-Transformer-Modul verarbeitet und dann an einen äquivarianten TFN-Transformer übergeben, der vorhergesagte Seitenkettenkoordinaten, Konfidenzwerte für jeden Rest und optionale Designsequenzen ausgibt. Die vorhergesagten Koordinaten werden nachbearbeitet, um alle räumlichen Konflikte zu beseitigen und eine idealisierte Geometrie sicherzustellen. In Bezug auf die Vorhersageleistung zeigt AttnPacker Verbesserungen in der Genauigkeit und Effizienz sowohl für natürliche als auch für nicht-natürliche Backbone-Strukturen. Gleichzeitig ist die physikalische Machbarkeit gewährleistet, Abweichungen von idealen Bindungslängen und -winkeln sind vernachlässigbar und es entsteht nur eine minimale atomare sterische Hinderung. Molecular Heart führt Vergleichstests mit AttnPacker und den aktuellen hochmodernen Methoden – SCWRL4, FASPR, RosettaPacker und DLPacker – an den natürlichen und nicht-nativen Protein-Backbone-Datensätzen CASP13 und CASP14 durch. Die Ergebnisse zeigen, dass AttnPacker herkömmliche Methoden zur Vorhersage von Proteinseitenketten auf nativen CASP13- und CASP14-Backbones deutlich übertrifft, wobei die durchschnittlichen Rekonstruktions-RMSDs bei jedem Testsatz um mehr als 18 % niedriger sind als bei der suboptimalen Methode. AttnPacker übertrifft auch die Deep-Learning-Methode DLPacker, indem es den durchschnittlichen RMSD um mehr als 11 % reduziert und gleichzeitig die Genauigkeit der Sidechain-Dieder deutlich verbessert. Zusätzlich zur Genauigkeit weist AttnPacker deutlich weniger Atomkollisionen auf als andere Methoden. Die Ergebnisse der Vorhersage der Seitenkettenstruktur jedes Algorithmus für die CASP13- und CASP14-Zielproteine, wenn die natürliche Hauptkettenstruktur angegeben ist. Sternchen zeigen an, dass die durchschnittlichen Konfliktwerte niedriger sind als die native Struktur – 56,0, 5,9 und 0,4 für CASP13 und 80,4, 7,9 und 2,5 für CASP14. Auf nicht-nativen CASP13- und CASP14-Backbones ist AttnPacker auch deutlich besser als andere Methoden, und die atomaren Kollisionen sind auch deutlich geringer als bei anderen Methoden. Die Ergebnisse der Vorhersage der Seitenkettenstruktur jedes Algorithmus für die CASP13- und CASP14-Zielproteine, wenn die nicht-natürliche Grundgerüststruktur angegeben ist. Sternchen zeigen an, dass die durchschnittlichen Konfliktwerte niedriger sind als die entsprechenden nativen Strukturen – 34,6, 2,2, 0,5 für CASP13 und 40,0, 2,7, 0,7 für CASP14. Innovativer Verzicht auf diskrete Rotamer-Bibliotheken und rechenintensive Konformationssuch- und Probenahmeschritte und direkte Kombination der 3D-Geometrie der Hauptkette, um alle Seitenkettenkoordinaten parallel zu berechnen. Im Vergleich zu der auf Deep Learning basierenden Methode DLPacker und der auf traditionellen Computermethoden basierenden RosettaPacker hat AttnPacker die Recheneffizienz deutlich verbessert und die Inferenzzeit um mehr als das Hundertfache verkürzt. Zeitvergleich verschiedener PSCP-Methoden. Rekonstruktion der relativen Zeiten der Seitenkettenatome für alle 83 CASP13-Zielproteine. AttnPacker schneidet beim Proteindesign ebenso gut ab. Molecular Heart hat eine AttnPacker-Variante für das Co-Design trainiert, die native Sequenzwiederherstellungsraten erreicht, die mit aktuellen Methoden auf dem neuesten Stand der Technik vergleichbar sind, und gleichzeitig hochpräzise Baugruppen produziert. Die Validierung der Rosetta-Simulation zeigt, dass von AttnPacker entworfene Strukturen im Allgemeinen subnative (niedrigere) Rosetta-Energien erzeugen. Vergleich von nativen Proteinsequenzen und von AttnPacker generierten Sequenzen unter Verwendung der ESMFold scTM- und plDDT-Metriken zur Bewertung der Qualität der AttnPacker-Generierung. Die Ergebnisse zeigten eine starke Korrelation. Zusätzlich zu seiner erstaunlichen Effektivität und Effizienz hat AttnPaker auch einen sehr praktischen Wert – es ist sehr einfach zu bedienen. Für die Ausführung von AttnPaker ist lediglich eine Proteinstrukturdatei erforderlich. Im Gegensatz dazu erfordert OPUS-Rota4 (28) eine Voxeldarstellung der atomaren Umgebung von DLPacker, Logik, Sekundärstruktur von trRosetta100 und Einschränkungsdateien von der OPUS-CM-Ausgabe. Da AttnPacker außerdem Seitenkettenkoordinaten direkt vorhersagt, ist die Ausgabe vollständig differenzierbar, was nachgelagerte Vorhersageaufgaben wie Optimierung oder Protein-Protein-Wechselwirkungen erleichtert. „Die Vorteile des guten Vorhersageeffekts, der hohen Effizienz und der Benutzerfreundlichkeit begünstigen den weit verbreiteten Einsatz von AttnPacker in Forschungs- und Industriebereichen“, sagte Professor Xu Jinbo. 1. AttnPacker ist ein SE (3)-Äquivariantenmodell, das zur direkten Vorhersage der Proteinseitenkettenstruktur und des Proteinsequenzdesigns verwendet werden kann arbeiten. 2. Die Genauigkeit von AttnPacker ist besser als bei anderen Methoden, die Effizienz ist erheblich verbessert und die Verwendung ist äußerst einfach. Effekt
Zusammenfassung
Das obige ist der detaillierte Inhalt vonWeltneuheit: Der neue Open-Source-KI-Algorithmus von Molecular Heart zur Überwindung der Probleme der Proteinseitenkettenvorhersage und des Sequenzdesigns. Für weitere Informationen folgen Sie bitte anderen verwandten Artikeln auf der PHP chinesischen Website!

Heiße KI -Werkzeuge

Undresser.AI Undress
KI-gestützte App zum Erstellen realistischer Aktfotos

AI Clothes Remover
Online-KI-Tool zum Entfernen von Kleidung aus Fotos.

Undress AI Tool
Ausziehbilder kostenlos

Clothoff.io
KI-Kleiderentferner

AI Hentai Generator
Erstellen Sie kostenlos Ai Hentai.

Heißer Artikel
Heiße Werkzeuge

Notepad++7.3.1
Einfach zu bedienender und kostenloser Code-Editor

SublimeText3 chinesische Version
Chinesische Version, sehr einfach zu bedienen

Senden Sie Studio 13.0.1
Leistungsstarke integrierte PHP-Entwicklungsumgebung

Dreamweaver CS6
Visuelle Webentwicklungstools

SublimeText3 Mac-Version
Codebearbeitungssoftware auf Gottesniveau (SublimeText3)
Heiße Themen
 1384
1384
 52
52

 CentOS Shutdown -Befehlszeile
Apr 14, 2025 pm 09:12 PM
CentOS Shutdown -Befehlszeile
Apr 14, 2025 pm 09:12 PM
Der Befehl centOS stilldown wird heruntergefahren und die Syntax wird von [Optionen] ausgeführt [Informationen]. Zu den Optionen gehören: -h das System sofort stoppen; -P schalten Sie die Leistung nach dem Herunterfahren aus; -r neu starten; -t Wartezeit. Zeiten können als unmittelbar (jetzt), Minuten (Minuten) oder als bestimmte Zeit (HH: MM) angegeben werden. Hinzugefügten Informationen können in Systemmeldungen angezeigt werden.
 So überprüfen Sie die CentOS -HDFS -Konfiguration
Apr 14, 2025 pm 07:21 PM
So überprüfen Sie die CentOS -HDFS -Konfiguration
Apr 14, 2025 pm 07:21 PM
Vollständige Anleitung zur Überprüfung der HDFS -Konfiguration in CentOS -Systemen In diesem Artikel wird die Konfiguration und den laufenden Status von HDFS auf CentOS -Systemen effektiv überprüft. Die folgenden Schritte helfen Ihnen dabei, das Setup und den Betrieb von HDFs vollständig zu verstehen. Überprüfen Sie die Hadoop -Umgebungsvariable: Stellen Sie zunächst sicher, dass die Hadoop -Umgebungsvariable korrekt eingestellt ist. Führen Sie im Terminal den folgenden Befehl aus, um zu überprüfen, ob Hadoop ordnungsgemäß installiert und konfiguriert ist: Hadoopsion-Check HDFS-Konfigurationsdatei: Die Kernkonfigurationsdatei von HDFS befindet sich im/etc/hadoop/conf/verzeichnis, wobei core-site.xml und hdfs-site.xml von entscheidender Bedeutung sind. verwenden
 Was sind die Backup -Methoden für Gitlab auf CentOS?
Apr 14, 2025 pm 05:33 PM
Was sind die Backup -Methoden für Gitlab auf CentOS?
Apr 14, 2025 pm 05:33 PM
Backup- und Wiederherstellungsrichtlinie von GitLab im Rahmen von CentOS -System Um die Datensicherheit und Wiederherstellung der Daten zu gewährleisten, bietet GitLab on CentOS eine Vielzahl von Sicherungsmethoden. In diesem Artikel werden mehrere gängige Sicherungsmethoden, Konfigurationsparameter und Wiederherstellungsprozesse im Detail eingeführt, um eine vollständige GitLab -Sicherungs- und Wiederherstellungsstrategie aufzubauen. 1. Manuell Backup Verwenden Sie den GitLab-RakegitLab: Backup: Befehl erstellen, um die manuelle Sicherung auszuführen. Dieser Befehl unterstützt wichtige Informationen wie GitLab Repository, Datenbank, Benutzer, Benutzergruppen, Schlüssel und Berechtigungen. Die Standardsicherungsdatei wird im Verzeichnis/var/opt/gitlab/backups gespeichert. Sie können /etc /gitlab ändern
 Wie ist die GPU -Unterstützung für Pytorch bei CentOS?
Apr 14, 2025 pm 06:48 PM
Wie ist die GPU -Unterstützung für Pytorch bei CentOS?
Apr 14, 2025 pm 06:48 PM
Aktivieren Sie die Pytorch -GPU -Beschleunigung am CentOS -System erfordert die Installation von CUDA-, CUDNN- und GPU -Versionen von Pytorch. Die folgenden Schritte führen Sie durch den Prozess: Cuda und Cudnn Installation Bestimmen Sie die CUDA-Version Kompatibilität: Verwenden Sie den Befehl nvidia-smi, um die von Ihrer NVIDIA-Grafikkarte unterstützte CUDA-Version anzuzeigen. Beispielsweise kann Ihre MX450 -Grafikkarte CUDA11.1 oder höher unterstützen. Download und installieren Sie Cudatoolkit: Besuchen Sie die offizielle Website von Nvidiacudatoolkit und laden Sie die entsprechende Version gemäß der höchsten CUDA -Version herunter und installieren Sie sie, die von Ihrer Grafikkarte unterstützt wird. Installieren Sie die Cudnn -Bibliothek:
 Detaillierte Erklärung des Docker -Prinzips
Apr 14, 2025 pm 11:57 PM
Detaillierte Erklärung des Docker -Prinzips
Apr 14, 2025 pm 11:57 PM
Docker verwendet Linux -Kernel -Funktionen, um eine effiziente und isolierte Anwendungsumgebung zu bieten. Sein Arbeitsprinzip lautet wie folgt: 1. Der Spiegel wird als schreibgeschützte Vorlage verwendet, die alles enthält, was Sie für die Ausführung der Anwendung benötigen. 2. Das Union File System (UnionFS) stapelt mehrere Dateisysteme, speichert nur die Unterschiede, speichert Platz und beschleunigt. 3. Der Daemon verwaltet die Spiegel und Container, und der Kunde verwendet sie für die Interaktion. 4. Namespaces und CGroups implementieren Container -Isolation und Ressourcenbeschränkungen; 5. Mehrere Netzwerkmodi unterstützen die Containerverbindung. Nur wenn Sie diese Kernkonzepte verstehen, können Sie Docker besser nutzen.
 CentOS installieren MySQL
Apr 14, 2025 pm 08:09 PM
CentOS installieren MySQL
Apr 14, 2025 pm 08:09 PM
Die Installation von MySQL auf CentOS umfasst die folgenden Schritte: Hinzufügen der entsprechenden MySQL Yum -Quelle. Führen Sie den Befehl mySQL-server aus, um den MySQL-Server zu installieren. Verwenden Sie den Befehl mySQL_SECURE_INSTALLATION, um Sicherheitseinstellungen vorzunehmen, z. B. das Festlegen des Stammbenutzerkennworts. Passen Sie die MySQL -Konfigurationsdatei nach Bedarf an. Tune MySQL -Parameter und optimieren Sie Datenbanken für die Leistung.
 So sehen Sie sich Gitlab -Protokolle unter CentOS
Apr 14, 2025 pm 06:18 PM
So sehen Sie sich Gitlab -Protokolle unter CentOS
Apr 14, 2025 pm 06:18 PM
Eine vollständige Anleitung zum Anzeigen von GitLab -Protokollen unter CentOS -System In diesem Artikel wird in diesem Artikel verschiedene GitLab -Protokolle im CentOS -System angezeigt, einschließlich Hauptprotokolle, Ausnahmebodi und anderen zugehörigen Protokollen. Bitte beachten Sie, dass der Log -Dateipfad je nach GitLab -Version und Installationsmethode variieren kann. Wenn der folgende Pfad nicht vorhanden ist, überprüfen Sie bitte das GitLab -Installationsverzeichnis und die Konfigurationsdateien. 1. Zeigen Sie das Hauptprotokoll an. Verwenden Sie den folgenden Befehl, um die Hauptprotokolldatei der GitLabRails-Anwendung anzuzeigen: Befehl: Sudocat/var/log/gitlab/gitlab-rails/production.log Dieser Befehl zeigt das Produkt an
 Wie man eine verteilte Schulung von Pytorch auf CentOS betreibt
Apr 14, 2025 pm 06:36 PM
Wie man eine verteilte Schulung von Pytorch auf CentOS betreibt
Apr 14, 2025 pm 06:36 PM
Pytorch Distributed Training on CentOS -System erfordert die folgenden Schritte: Pytorch -Installation: Die Prämisse ist, dass Python und PIP im CentOS -System installiert sind. Nehmen Sie abhängig von Ihrer CUDA -Version den entsprechenden Installationsbefehl von der offiziellen Pytorch -Website ab. Für CPU-Schulungen können Sie den folgenden Befehl verwenden: PipinstallTorChTorChVisionTorChaudio Wenn Sie GPU-Unterstützung benötigen, stellen Sie sicher, dass die entsprechende Version von CUDA und CUDNN installiert ist und die entsprechende Pytorch-Version für die Installation verwenden. Konfiguration der verteilten Umgebung: Verteiltes Training erfordert in der Regel mehrere Maschinen oder mehrere Maschinen-Mehrfach-GPUs. Ort
