
 Technologie-Peripheriegeräte
KI
KI-Arzneimittelforscher tritt der Unterzeitschrift „Nature' bei: Nutzung von Fachwissen zur Beschleunigung der Arzneimittelentwicklung
Technologie-Peripheriegeräte
KI
KI-Arzneimittelforscher tritt der Unterzeitschrift „Nature' bei: Nutzung von Fachwissen zur Beschleunigung der Arzneimittelentwicklung
KI-Arzneimittelforscher tritt der Unterzeitschrift „Nature' bei: Nutzung von Fachwissen zur Beschleunigung der Arzneimittelentwicklung

Die Entdeckung von Arzneimitteln ist ein komplexer, mehrstufiger Prozess, der die Schnittstelle vieler Teildisziplinen der Chemie und Biologie umfasst. Humanmedizinische Chemiker spielen dabei mit ihrer jahrelang gesammelten Expertise eine wichtige Rolle
Kann also künstliche Intelligenz (KI) die Rolle übernehmen, die medizinische Chemiker bei der Arzneimittelentwicklung spielen? Die Antwort könnte ja sein.
Kürzlich hat ein Forschungsteam der Novartis Institutes for Biomedical Research (NIBR) und des Microsoft Research Center for Scientific Intelligence (AI4Science) gemeinsam ein Modell für maschinelles Lernen vorgeschlagen, das das von professionellen Chemikern in ihrer Arbeit gesammelte kollektive Wissen teilweise reproduzieren kann Diese Art von Wissen wird oft als „chemische Intuition“ bezeichnet.
Das Forschungsteam ist davon überzeugt, dass diese Methode als Ergänzung zur molekularen Modellierung eingesetzt werden kann, um die Effizienz der zukünftigen Arzneimittelentwicklung zu verbessern
Die Forschungsarbeit trägt den Titel „Extracting Intuition in Medicinal Chemistry through Preference Machine Learning“ und wurde in Nature Communications, einer Unterzeitschrift von Nature, veröffentlicht

Maschinelles Lernen stellt das Fachwissen von Medizinchemikern wieder her
Medizinische Chemiker, sowohl Nasslabor- als auch Computerchemiker, spielen eine entscheidende Rolle in der Phase der „Leitoptimierung“ der Arzneimittelentwicklung, da sie oft gebeten werden, zu bestimmen, welche Verbindungen synthetisiert und in nachfolgenden Optimierungsrunden evaluiert werden müssen.Zu diesem Zweck überprüfen medizinische Chemiker typischerweise Daten, einschließlich der Eigenschaften von Verbindungen wie Aktivität, ADMET2 oder Informationen zur Zielstruktur. Daher hängt der Erfolg eines Projekts nicht nur von der Qualität der generierten experimentellen Daten ab, sondern auch von der Robustheit und Rationalität der Entscheidungen des in der medizinischen Chemie arbeitenden Teams.
Medizinische Chemiker können Entscheidungen effizienter treffen, da sie häufig auf Fachwissen zurückgreifen, um ein intuitives Verständnis dafür zu entwickeln, was in verschiedenen Iterationen der frühen Arzneimittelforschung gelingt.
Während es bereits frühere Versuche gab, dieses Wissen mithilfe regelbasierter Ansätze oder einfacher chemoinformatischer Machbarkeitsbewertungen zu formalisieren, bleibt die Erfassung der Subtilität und Komplexität der Bewertung durch medizinische Chemiker eine grundlegende HerausforderungUm dieses Ziel zu erreichen, zielt die Forschung darauf ab, Fachwissen in einen Teil eines maschinellen Lernmodells umzuwandeln. Dieses Modell kann wie andere Empfehlungssysteme, über die in der Branche berichtet wurde, als Hilfswerkzeug verwendet werden, um den Entscheidungsprozess bei der Lead-Optimierung oder anderen Aspekten der Arzneimittelentwicklung einzusetzen
Wenn man bedenkt, dass die medizinische Chemie derzeit hauptsächlich auf Handarbeit beruht, ist sie zwangsläufig von subjektiven Voreingenommenheiten betroffen. Einige Studien haben eine geringe Übereinstimmung bei den Bewertungen sowohl zwischen medizinischen Chemikern als auch innerhalb medizinischer Chemiker festgestellt.
In dieser Studie hoffen die Forscher, einige Probleme zu lösen, indem sie Strategien aus Multiplayer-Spielen übernehmen.Sie behandelten die Aufgabe, eine Reihe von Molekülen einzuordnen, als Präferenz-Lernproblem und verwendeten dann ein einfaches neuronales Netzwerk, um individuelle Präferenzen zu modellieren.
Abbildung |. Gesamtschematische Darstellung der Hauptideen der Forschung (Quelle: das Papier)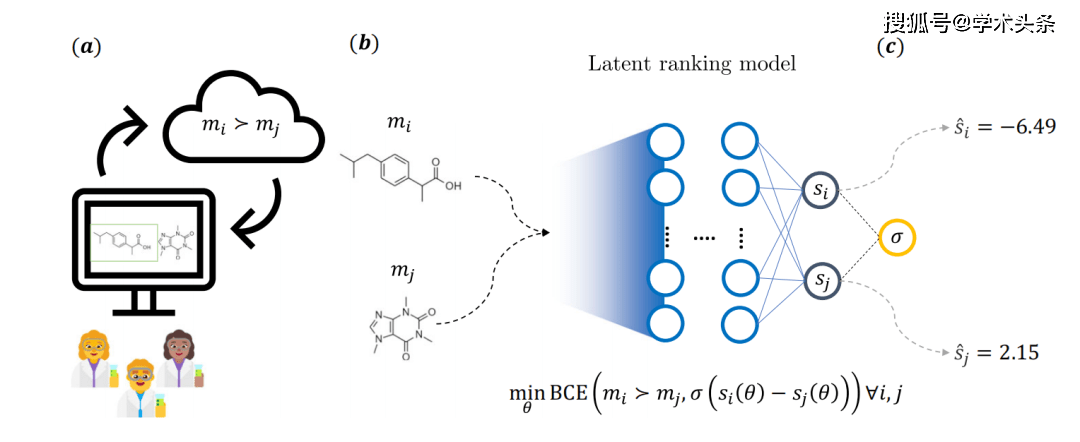 Konkret werden Moleküle, wie in der Abbildung oben dargestellt, als Teilnehmer eines Wettbewerbsspiels betrachtet, wobei die Wahrscheinlichkeit, dass eine Seite gewinnt, durch das Feedback des Chemikers bestimmt wird. Dazu beantworten medizinische Chemiker in einer Webanwendung vorgegebene Fragen und wählen eines von zwei Molekülen aus. Insgesamt waren 35 medizinische Chemiker von Novartis an dem Prozess beteiligt, der zur Sammlung von mehr als 5.000 Anmerkungen führte.
Konkret werden Moleküle, wie in der Abbildung oben dargestellt, als Teilnehmer eines Wettbewerbsspiels betrachtet, wobei die Wahrscheinlichkeit, dass eine Seite gewinnt, durch das Feedback des Chemikers bestimmt wird. Dazu beantworten medizinische Chemiker in einer Webanwendung vorgegebene Fragen und wählen eines von zwei Molekülen aus. Insgesamt waren 35 medizinische Chemiker von Novartis an dem Prozess beteiligt, der zur Sammlung von mehr als 5.000 Anmerkungen führte.
Dieses Feedback führte zu einem impliziten Bewertungsmodell, das ein Modell mit zwei unabhängigen neuronalen Netzwerkstrukturen verwendet. Jeder Zweig hat ein festes Gewicht und die Moleküle werden mithilfe gemeinsamer Chemoinformatik-Deskriptoren charakterisiert. Während des Trainings werden die Parameter des Modells über einen binären Kreuzentropieverlust (BCE-Verlust) optimiert, der von der zugrunde liegenden Bewertungsdifferenz eines Molekülpaares und dem Feedback des Chemikers abhängt
Sobald das Training abgeschlossen ist, kann die Punktzahl für jedes beliebige Molekül abgeleitet werden, die dann für nachgelagerte Aufgaben in der Chemieinformatik verwendet werden kann.
Darüber hinaus kann das Modell die Ähnlichkeiten zwischen verschiedenen Medikamenten genauer bestimmen. Die in der Studie vorgeschlagene Lernbewertungsfunktion ist genauer als der herkömmliche Arzneimittelähnlichkeitsbewertungsindex (QED)Insbesondere
Um die Reproduzierbarkeit der Studie und die Weiterentwicklung des Fachgebiets zu fördern, stellen die Forscher auch ein Softwarepaket namens „MolSkill“ zur Verfügung, dasdas Modell und anonymisierte Antwortdaten enthält.
Probleme und Anwendungen des maschinellen Lernens im Bereich der medizinischen ChemieObwohl dieses Modell das von medizinischen Chemikern in ihrer Arbeit gesammelte Wissen reproduzieren kann, weist es auch einige Einschränkungen auf. Um die chemische Intuition zu erfassen, waren die bei der Datenerfassung gestellten Fragen zunächst immer vage. Auch wenn das vorgeschlagene Studiendesign im Vergleich zu früheren Studien zu einer größeren Übereinstimmung zwischen den Teilnehmern führte, ist die Methode des paarweisen Vergleichs nicht perfekt. Darüber hinaus führt der „Flatland-Irrtum“ dazu, dass Menschen dazu neigen, hochdimensionale Probleme in einen kleinen Satz von Variablen zu vereinfachen, die kognitiv verfolgt werden können, und diese Vereinfachung kann durch die persönlichen Eigenschaften jedes medizinischen Chemikers beeinflusst werden Das Forschungsteam gab jedoch an, dass das in dieser Studie vorgeschlagene Modell nicht auf den Anwendungsbereich der aktuellen Studie beschränkt ist. Insbesondere kann der besprochene Rahmen auf andere quantifizierbare, aber teure Observablen im Bereich der Arzneimittelentwicklung ausgeweitet werden. Darüber hinaus kann es Einblicke in noch unerforschte Bereiche des chemischen Raums geben. Vor diesem Hintergrund glaubt das Forschungsteam, dass eine ähnliche Architektur aufgebaut werden kann, indem man einige beliebte regelbasierte Filter aus künstlich generierten Trainingsdaten lernen lässt. Dieses Modell kann die große Einschränkung überwinden, die darin besteht, dass Verbindungen manuell gefiltert werden müssen, bevor Schlussfolgerungen gezogen werden können Der gleiche Ansatz kann auch zur Generierung von Verbindungsbewertungen verwendet werden, indem Kombinationen in Bibliotheken synthetischer Chemikalien priorisiert werden, bei denen das Screening aufgrund ihrer natürlichen Neuheit mit vorhandenen regelbasierten Methoden schwierig ist Eine weitere Sache, die noch einmal zum Ausdruck gebracht werden muss, ist: In einem prospektiven, primären Optimierungsszenario für ein bestimmtes Ziel müssen mehrere Informationsquellen (wie biologische Eigenschaften, ADMET usw.) umfassend berücksichtigt werden, um die Praktikabilität des Ziels zu testen Forschungsrahmen Das Forschungsteam schrieb in der Arbeit: „Maschinelle Lernmethoden können Tausende von Verbindungen entwerfen, und Technologien wie das Hochdurchsatz-Screening können eine große Anzahl von Kandidatenverbindungen in den frühen Phasen des Arzneimittelentwicklungsprozesses hervorheben.“ Die Bewertungsmethode schlug dies vor Es wird erwartet, dass diese Anwendung in den kommenden Jahren die Akzeptanz und das Vertrauen der Methode beschleunigen wird
Das obige ist der detaillierte Inhalt vonKI-Arzneimittelforscher tritt der Unterzeitschrift „Nature' bei: Nutzung von Fachwissen zur Beschleunigung der Arzneimittelentwicklung. Für weitere Informationen folgen Sie bitte anderen verwandten Artikeln auf der PHP chinesischen Website!

Heiße KI -Werkzeuge

Undresser.AI Undress
KI-gestützte App zum Erstellen realistischer Aktfotos

AI Clothes Remover
Online-KI-Tool zum Entfernen von Kleidung aus Fotos.

Undress AI Tool
Ausziehbilder kostenlos

Clothoff.io
KI-Kleiderentferner

AI Hentai Generator
Erstellen Sie kostenlos Ai Hentai.

Heißer Artikel
Heiße Werkzeuge

Notepad++7.3.1
Einfach zu bedienender und kostenloser Code-Editor

SublimeText3 chinesische Version
Chinesische Version, sehr einfach zu bedienen

Senden Sie Studio 13.0.1
Leistungsstarke integrierte PHP-Entwicklungsumgebung

Dreamweaver CS6
Visuelle Webentwicklungstools

SublimeText3 Mac-Version
Codebearbeitungssoftware auf Gottesniveau (SublimeText3)
Heiße Themen
 1374
1374
 52
52

 Ich habe versucht, die Vibe -Codierung mit Cursor AI und es ist erstaunlich!
Mar 20, 2025 pm 03:34 PM
Ich habe versucht, die Vibe -Codierung mit Cursor AI und es ist erstaunlich!
Mar 20, 2025 pm 03:34 PM
Die Vibe -Codierung verändert die Welt der Softwareentwicklung, indem wir Anwendungen mit natürlicher Sprache anstelle von endlosen Codezeilen erstellen können. Inspiriert von Visionären wie Andrej Karpathy, lässt dieser innovative Ansatz Dev
 Top 5 Genai Starts vom Februar 2025: GPT-4,5, GROK-3 & MEHR!
Mar 22, 2025 am 10:58 AM
Top 5 Genai Starts vom Februar 2025: GPT-4,5, GROK-3 & MEHR!
Mar 22, 2025 am 10:58 AM
Februar 2025 war ein weiterer bahnbrechender Monat für die Generative KI, die uns einige der am meisten erwarteten Modell-Upgrades und bahnbrechenden neuen Funktionen gebracht hat. Von Xais Grok 3 und Anthropics Claude 3.7 -Sonett, um g zu eröffnen
 Wie benutze ich Yolo V12 zur Objekterkennung?
Mar 22, 2025 am 11:07 AM
Wie benutze ich Yolo V12 zur Objekterkennung?
Mar 22, 2025 am 11:07 AM
Yolo (Sie schauen nur einmal) war ein führender Echtzeit-Objekterkennungsrahmen, wobei jede Iteration die vorherigen Versionen verbessert. Die neueste Version Yolo V12 führt Fortschritte vor, die die Genauigkeit erheblich verbessern
 Sora vs Veo 2: Welches erstellt realistischere Videos?
Mar 10, 2025 pm 12:22 PM
Sora vs Veo 2: Welches erstellt realistischere Videos?
Mar 10, 2025 pm 12:22 PM
Google's Veo 2 und Openais Sora: Welcher AI -Videogenerator regiert oberste? Beide Plattformen erzeugen beeindruckende KI -Videos, aber ihre Stärken liegen in verschiedenen Bereichen. Dieser Vergleich unter Verwendung verschiedener Eingabeaufforderungen zeigt, welches Werkzeug Ihren Anforderungen am besten entspricht. T
 Gencast von Google: Wettervorhersage mit Gencast Mini Demo
Mar 16, 2025 pm 01:46 PM
Gencast von Google: Wettervorhersage mit Gencast Mini Demo
Mar 16, 2025 pm 01:46 PM
Gencast von Google Deepmind: Eine revolutionäre KI für die Wettervorhersage Die Wettervorhersage wurde einer dramatischen Transformation unterzogen, die sich von rudimentären Beobachtungen zu ausgefeilten AI-angetriebenen Vorhersagen überschreitet. Google DeepMinds Gencast, ein Bodenbrei
 Ist Chatgpt 4 o verfügbar?
Mar 28, 2025 pm 05:29 PM
Ist Chatgpt 4 o verfügbar?
Mar 28, 2025 pm 05:29 PM
Chatgpt 4 ist derzeit verfügbar und weit verbreitet, wodurch im Vergleich zu seinen Vorgängern wie ChatGPT 3.5 signifikante Verbesserungen beim Verständnis des Kontextes und des Generierens kohärenter Antworten zeigt. Zukünftige Entwicklungen können mehr personalisierte Inters umfassen
 Welche KI ist besser als Chatgpt?
Mar 18, 2025 pm 06:05 PM
Welche KI ist besser als Chatgpt?
Mar 18, 2025 pm 06:05 PM
Der Artikel erörtert KI -Modelle, die Chatgpt wie Lamda, Lama und Grok übertreffen und ihre Vorteile in Bezug auf Genauigkeit, Verständnis und Branchenauswirkungen hervorheben. (159 Charaktere)
 O1 gegen GPT-4O: Ist OpenAIs neues Modell besser als GPT-4O?
Mar 16, 2025 am 11:47 AM
O1 gegen GPT-4O: Ist OpenAIs neues Modell besser als GPT-4O?
Mar 16, 2025 am 11:47 AM
Openais O1: Ein 12-tägiger Geschenkbummel beginnt mit ihrem bisher mächtigsten Modell Die Ankunft im Dezember bringt eine globale Verlangsamung, Schneeflocken in einigen Teilen der Welt, aber Openai fängt gerade erst an. Sam Altman und sein Team starten ein 12-tägiges Geschenk Ex
