
 Technologie-Peripheriegeräte
KI
Die Peking-Universität und Wangshi Intelligence schlagen ein neues Modell vor: die Lücke zwischen dem Vortraining chemischer Reaktionen und der bedingten Molekülgenerierung zu schließen!
Technologie-Peripheriegeräte
KI
Die Peking-Universität und Wangshi Intelligence schlagen ein neues Modell vor: die Lücke zwischen dem Vortraining chemischer Reaktionen und der bedingten Molekülgenerierung zu schließen!
Die Peking-Universität und Wangshi Intelligence schlagen ein neues Modell vor: die Lücke zwischen dem Vortraining chemischer Reaktionen und der bedingten Molekülgenerierung zu schließen!

Chemische Reaktionen sind die Grundlage des Arzneimitteldesigns und der Forschung in der organischen Chemie. In der Forschungsgemeinschaft besteht ein wachsender Bedarf an einem groß angelegten Deep-Learning-Framework, das die grundlegenden Regeln chemischer Reaktionen effektiv erfassen kann.
Kürzlich hat ein Forschungsteam der Peking-Universität und Wangshi Intelligence eine neue Methode vorgeschlagen, um die Lücke zwischen reaktionsbasiertem molekularem Vortraining und Generierungsaufgaben zu schließen.
Inspiriert von den Mechanismen der organischen Chemie haben Forscher ein neues Pre-Training-Framework entwickelt, das es ermöglicht, induktive Voreingenommenheit in Modelle zu integrieren. Dieses vorgeschlagene Framework erzielt Ergebnisse auf dem neuesten Stand der Technik bei der Durchführung anspruchsvoller nachgelagerter Aufgaben. Durch die Nutzung chemischer Kenntnisse überwindet das Framework die Einschränkungen aktueller Modelle zur Molekülerzeugung, die auf einer kleinen Anzahl von Reaktionsvorlagen basieren. In umfangreichen Experimenten generierte das Modell hochwertige, synthetisierbare wirkstoffähnliche Strukturen
Insgesamt ist diese Forschung ein wichtiger Schritt hin zu einem groß angelegten Deep-Learning-Framework für eine Vielzahl reaktionsbasierter Anwendungen.
Die Studie trug den Titel „Überbrückung der Lücke zwischen chemischem Reaktions-Pretraining und bedingter Molekülgenerierung mit einem einheitlichen Modell“ und wurde am 5. Dezember 2023 in „Nature Machine Intelligence“ veröffentlicht.

Link zum Papier: https://www.nature.com/articles/s42256-023-00764-9
Deep-Learning-Modelle werden in vielen wissenschaftlichen Forschungsbereichen häufig eingesetzt. Das Pre-Training-Framework spielt eine positive Rolle bei der nahtlosen Integration neuer Aufgaben und kann den Modellierungsprozess beschleunigen, insbesondere wenn die markierten Daten begrenzt sind.
Die Grundlage des Arzneimitteldesigns und der Forschung in der organischen Chemie sind chemische Reaktionen. Derzeit haben die Forschung und Anwendung von Data Mining den Einsatz von Deep-Learning-Modellen in chemischen Reaktionen ermöglicht. Basierend auf diesen Daten gab es viele datengesteuerte Studien, die sich mit dem Repräsentationslernen chemischer Reaktionen befassten.
Repräsentationslernen bezieht sich auf das automatische Lernen nützlicher Funktionen aus Daten und deren anschließende Verwendung für verschiedene nachgelagerte Aufgaben. Bestehende Methoden ignorieren grundlegende Theorien der organischen Chemie und schränken ihre Leistung ein.
Molekülgenerierung auf Basis chemischer Reaktionen
Neben der Reaktionsklassifizierungsaufgabe ist auch die Molekülgenerierung auf Basis chemischer Reaktionen eine wichtige Anwendung. In frühen Studien wurden häufig templatbasierte Strategien zur schrittweisen Molekülgenerierung übernommen
Diese templatbasierten Methoden stützten sich stark auf vordefinierte Bausteine und Reaktionen, was den zugänglichen chemischen Raum einschränkte. Ein ähnlicher Trend ist im Bereich der Reaktionsproduktvorhersage zu beobachten, wo templatbasierte Methoden nicht auf komplexe Reaktionen übertragen werden können; dieses Problem kann durch den Einsatz templatfreier Methoden gelöst werden.
Bei reaktionsbasierten Molekülgenerierungsaufgaben zeigen templatfreie Methoden auch Generalisierungsvorteile gegenüber templatbasierten Methoden. Allerdings können bestehende templatfreie Methoden zur Molekülgenerierung nur Moleküle erzeugen, die auf vordefinierten Reaktantenbibliotheken basieren. Darüber hinaus ist es für die Leitverbindungs- oder Leitstrukturoptimierungsphase im Arzneimitteldesign vorteilhafter, chemische Reaktionen als Bearbeitungswerkzeuge zu nutzen, um eine bestimmte Struktur zu modifizieren. Die resultierende chemische Bibliothek wird sich auf einen Teilbereich des chemischen Raums konzentrieren, der mit weniger Reaktionsschritten synthetisiert werden kann.
Ein neues, umfassendes Deep-Learning-Framework für chemische Reaktionen
Hier schlagen Forscher ein neues, umfassendes Deep-Learning-Framework für chemische Reaktionen vor, genannt Uni-RXN. Ziel ist es, zwei grundlegende Aufgaben zu lösen: selbstüberwachtes Repräsentationslernen und bedingte generative Modellierung.
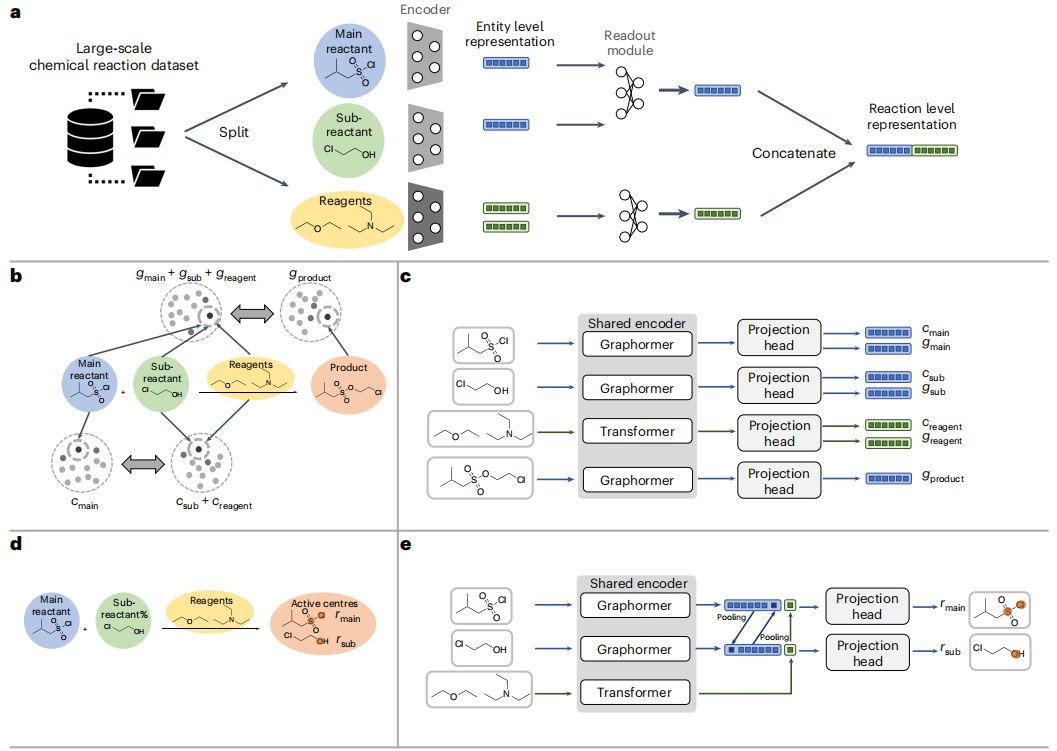
Anzeigen: Zusammensetzung und Methode von Uni-RXN. (Zitiert aus: Artikel)
Anders als bestehende Methoden schlugen die Forscher eine Reihe selbstüberwachter Aufgaben vor, die speziell für chemische Reaktionen entwickelt wurden. Zu diesen Aufgaben gehören die Vorhersage des Reaktionszentrums, die Paarung von Primärreaktanten und Unterreaktanten sowie die Paarung von Reaktanten und Produkten. In einer umfassenden Bewertung anspruchsvoller Reaktionsaufgaben übertrifft die Uni-RXN-Methode den Stand der Technik und demonstriert ihre Fähigkeit, Domänenwissen über chemische Reaktionen effektiv zu erfassen. Die erzielten vielversprechenden Ergebnisse ebnen den Weg für weitreichende nachgelagerte Anwendungen
Durch die effektive Erfassung chemischer Regeln ist Uni-RXN ideal für Erzeugungsaufgaben geeignet. Im Gegensatz zu herkömmlichen Methoden, die auf der Auswahl von Fragmenten aus einer Bibliothek vordefinierter Reaktanten basieren, verwendet Uni-RXN molekulare Strukturen als Eingabebedingungen und generiert Darstellungen der entsprechenden Reaktanten, während die Permutationsinvarianz innerhalb der Reaktion erhalten bleibt. Uni-RXN nutzt die Leistungsfähigkeit des Suchpakets für dichte Vektorähnlichkeiten und ermöglicht die effiziente Suche nach Reaktanten aus großen Reaktanten- und Reagenzienbibliotheken. Anschließend wird ein Reaktionsvorhersagemodell verwendet, um Produktergebnisse zu generieren.
Im Vergleich zu templatbasierten Methoden, die nur einen begrenzten Teilbereich des chemischen Raums erforschen, weist Uni-RXN eine überlegene Leistung bei der Erzeugung eines breiteren Spektrums synthetisierbarer arzneimittelähnlicher Strukturen auf. Aufgrund dieser Funktion eignet es sich besonders für die Aufzählung virtueller Bibliotheken und wird durch umfassende statistische Analysen und Fallstudien unterstützt.
Die Uni-RXN-Methode bietet viele Vorteile und kann umfangreiche Darstellungen für die anspruchsvolle Aufgabe der Klassifizierung chemischer Reaktionen generieren. Im Vergleich zu anderen Basismodellen erreicht Uni-RXN eine Genauigkeit von 58,7 % mit nur 4 Datenpunkten pro Klasse. Umgeschriebener Inhalt: Die Genauigkeit der Klassifizierung chemischer Reaktionen ist in Tabelle 1 dargestellt. (Quelle: Paper)
Transformer-Modell kann verwendet werden, um zwischen optimierten und nicht optimierten chemischen Reaktionsdaten zu unterscheiden. Darüber hinaus kann der Encoder auch problemlos auf die Generierung struktureller Bedingungen angewendet werden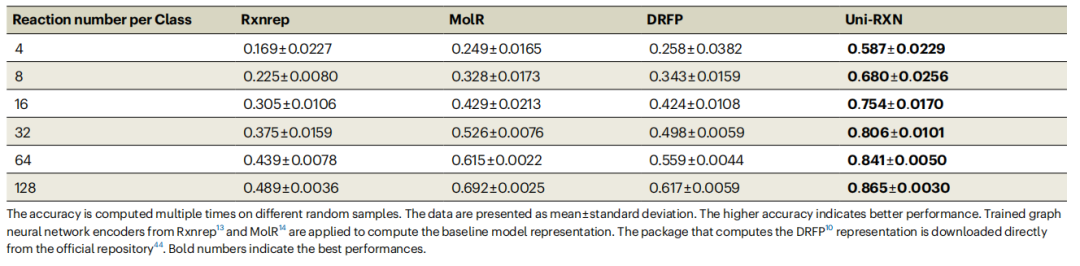
Der Inhalt, der neu geschrieben werden muss, ist: Das Diagramm zeigt die Abrufleistung und das Aufmerksamkeitsgewicht von Uni-RXN. (Quelle: Papier) 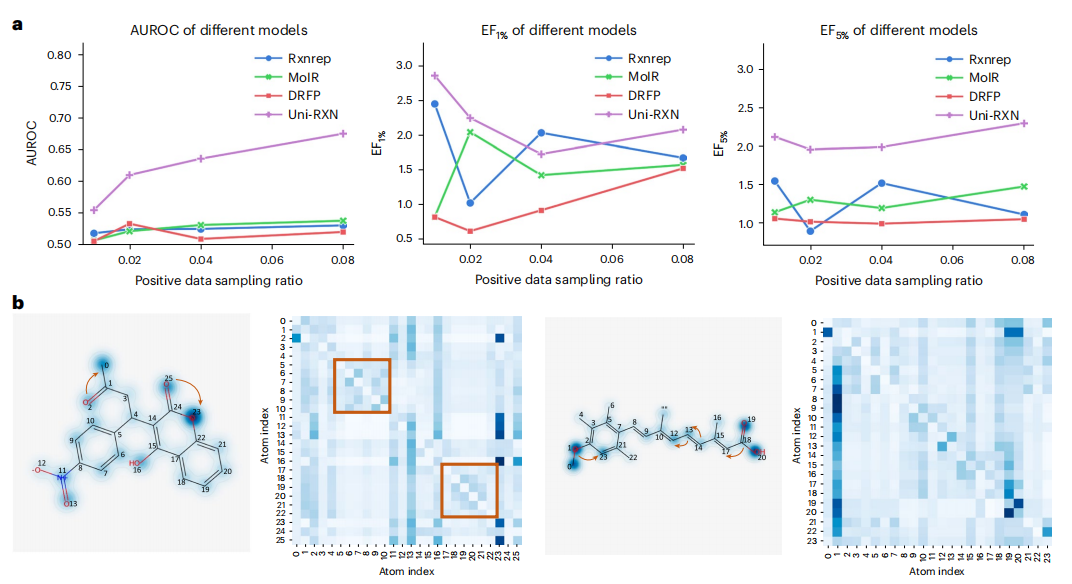
Die Ergebnisse verdeutlichen die günstigen Eigenschaften der durch das vorgeschlagene Modell erzeugten Moleküle, die sie für Aufgaben der Arzneimittelforschung gut geeignet machen. Dieses Modell kann mehr Moleküle mit arzneimittelähnlichen Eigenschaften und Synthetisierbarkeit erzeugen
Abbildung: Uni-RXNGen-Prozess und Leistung. (Quelle: Papier) 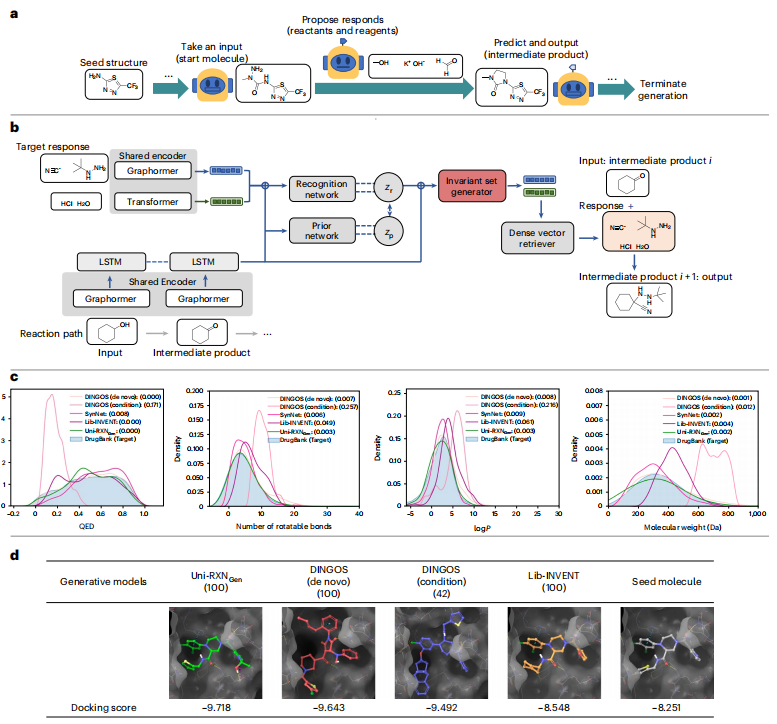
In Kombination mit virtuellen Screening-Methoden wie Molecular Docking kann dieses generierte Modell eine effiziente Struktur-Aktivitäts-Beziehungsforschung ermöglichen. Der riesige, durch dieses Modell erzeugte Raum für synthetische drogenähnliche Chemikalien kann die tatsächliche positive Rate bei der Wiederverwendung von Medikamenten oder bei der Suche nach Treffermolekülen verbessern.
Das obige ist der detaillierte Inhalt vonDie Peking-Universität und Wangshi Intelligence schlagen ein neues Modell vor: die Lücke zwischen dem Vortraining chemischer Reaktionen und der bedingten Molekülgenerierung zu schließen!. Für weitere Informationen folgen Sie bitte anderen verwandten Artikeln auf der PHP chinesischen Website!

Heiße KI -Werkzeuge

Undresser.AI Undress
KI-gestützte App zum Erstellen realistischer Aktfotos

AI Clothes Remover
Online-KI-Tool zum Entfernen von Kleidung aus Fotos.

Undress AI Tool
Ausziehbilder kostenlos

Clothoff.io
KI-Kleiderentferner

AI Hentai Generator
Erstellen Sie kostenlos Ai Hentai.

Heißer Artikel
Heiße Werkzeuge

Notepad++7.3.1
Einfach zu bedienender und kostenloser Code-Editor

SublimeText3 chinesische Version
Chinesische Version, sehr einfach zu bedienen

Senden Sie Studio 13.0.1
Leistungsstarke integrierte PHP-Entwicklungsumgebung

Dreamweaver CS6
Visuelle Webentwicklungstools

SublimeText3 Mac-Version
Codebearbeitungssoftware auf Gottesniveau (SublimeText3)
Heiße Themen
 1377
1377
 52
52

 Das weltweit leistungsstärkste Open-Source-MoE-Modell ist da, mit chinesischen Fähigkeiten, die mit GPT-4 vergleichbar sind, und der Preis beträgt nur fast ein Prozent von GPT-4-Turbo
May 07, 2024 pm 04:13 PM
Das weltweit leistungsstärkste Open-Source-MoE-Modell ist da, mit chinesischen Fähigkeiten, die mit GPT-4 vergleichbar sind, und der Preis beträgt nur fast ein Prozent von GPT-4-Turbo
May 07, 2024 pm 04:13 PM
Stellen Sie sich ein Modell der künstlichen Intelligenz vor, das nicht nur die Fähigkeit besitzt, die traditionelle Datenverarbeitung zu übertreffen, sondern auch eine effizientere Leistung zu geringeren Kosten erzielt. Dies ist keine Science-Fiction, DeepSeek-V2[1], das weltweit leistungsstärkste Open-Source-MoE-Modell, ist da. DeepSeek-V2 ist ein leistungsstarkes MoE-Sprachmodell (Mix of Experts) mit den Merkmalen eines wirtschaftlichen Trainings und einer effizienten Inferenz. Es besteht aus 236B Parametern, von denen 21B zur Aktivierung jedes Markers verwendet werden. Im Vergleich zu DeepSeek67B bietet DeepSeek-V2 eine stärkere Leistung, spart gleichzeitig 42,5 % der Trainingskosten, reduziert den KV-Cache um 93,3 % und erhöht den maximalen Generierungsdurchsatz auf das 5,76-fache. DeepSeek ist ein Unternehmen, das sich mit allgemeiner künstlicher Intelligenz beschäftigt
 KI untergräbt die mathematische Forschung! Der Gewinner der Fields-Medaille und der chinesisch-amerikanische Mathematiker führten 11 hochrangige Arbeiten an | Gefällt mir bei Terence Tao
Apr 09, 2024 am 11:52 AM
KI untergräbt die mathematische Forschung! Der Gewinner der Fields-Medaille und der chinesisch-amerikanische Mathematiker führten 11 hochrangige Arbeiten an | Gefällt mir bei Terence Tao
Apr 09, 2024 am 11:52 AM
KI verändert tatsächlich die Mathematik. Vor kurzem hat Tao Zhexuan, der diesem Thema große Aufmerksamkeit gewidmet hat, die neueste Ausgabe des „Bulletin of the American Mathematical Society“ (Bulletin der American Mathematical Society) weitergeleitet. Zum Thema „Werden Maschinen die Mathematik verändern?“ äußerten viele Mathematiker ihre Meinung. Der gesamte Prozess war voller Funken, knallhart und aufregend. Der Autor verfügt über eine starke Besetzung, darunter der Fields-Medaillengewinner Akshay Venkatesh, der chinesische Mathematiker Zheng Lejun, der NYU-Informatiker Ernest Davis und viele andere bekannte Wissenschaftler der Branche. Die Welt der KI hat sich dramatisch verändert. Viele dieser Artikel wurden vor einem Jahr eingereicht.
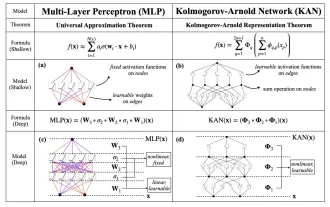 KAN, das MLP ersetzt, wurde durch Open-Source-Projekte auf Faltung erweitert
Jun 01, 2024 pm 10:03 PM
KAN, das MLP ersetzt, wurde durch Open-Source-Projekte auf Faltung erweitert
Jun 01, 2024 pm 10:03 PM
Anfang dieses Monats schlugen Forscher des MIT und anderer Institutionen eine vielversprechende Alternative zu MLP vor – KAN. KAN übertrifft MLP in Bezug auf Genauigkeit und Interpretierbarkeit. Und es kann MLP, das mit einer größeren Anzahl von Parametern ausgeführt wird, mit einer sehr kleinen Anzahl von Parametern übertreffen. Beispielsweise gaben die Autoren an, dass sie KAN nutzten, um die Ergebnisse von DeepMind mit einem kleineren Netzwerk und einem höheren Automatisierungsgrad zu reproduzieren. Konkret verfügt DeepMinds MLP über etwa 300.000 Parameter, während KAN nur etwa 200 Parameter hat. KAN hat eine starke mathematische Grundlage wie MLP und basiert auf dem universellen Approximationssatz, während KAN auf dem Kolmogorov-Arnold-Darstellungssatz basiert. Wie in der folgenden Abbildung gezeigt, hat KAN
 Hallo, elektrischer Atlas! Der Boston Dynamics-Roboter erwacht wieder zum Leben, seltsame 180-Grad-Bewegungen machen Musk Angst
Apr 18, 2024 pm 07:58 PM
Hallo, elektrischer Atlas! Der Boston Dynamics-Roboter erwacht wieder zum Leben, seltsame 180-Grad-Bewegungen machen Musk Angst
Apr 18, 2024 pm 07:58 PM
Boston Dynamics Atlas tritt offiziell in die Ära der Elektroroboter ein! Gestern hat sich der hydraulische Atlas einfach „unter Tränen“ von der Bühne der Geschichte zurückgezogen. Heute gab Boston Dynamics bekannt, dass der elektrische Atlas im Einsatz ist. Es scheint, dass Boston Dynamics im Bereich kommerzieller humanoider Roboter entschlossen ist, mit Tesla zu konkurrieren. Nach der Veröffentlichung des neuen Videos wurde es innerhalb von nur zehn Stunden bereits von mehr als einer Million Menschen angesehen. Die alten Leute gehen und neue Rollen entstehen. Das ist eine historische Notwendigkeit. Es besteht kein Zweifel, dass dieses Jahr das explosive Jahr der humanoiden Roboter ist. Netizens kommentierten: Die Weiterentwicklung der Roboter hat dazu geführt, dass die diesjährige Eröffnungsfeier wie Menschen aussieht, und der Freiheitsgrad ist weitaus größer als der von Menschen. Aber ist das wirklich kein Horrorfilm? Zu Beginn des Videos liegt Atlas ruhig auf dem Boden, scheinbar auf dem Rücken. Was folgt, ist atemberaubend
 Google ist begeistert: JAX-Leistung übertrifft Pytorch und TensorFlow! Es könnte die schnellste Wahl für das GPU-Inferenztraining werden
Apr 01, 2024 pm 07:46 PM
Google ist begeistert: JAX-Leistung übertrifft Pytorch und TensorFlow! Es könnte die schnellste Wahl für das GPU-Inferenztraining werden
Apr 01, 2024 pm 07:46 PM
Die von Google geförderte Leistung von JAX hat in jüngsten Benchmark-Tests die von Pytorch und TensorFlow übertroffen und belegt bei 7 Indikatoren den ersten Platz. Und der Test wurde nicht auf der TPU mit der besten JAX-Leistung durchgeführt. Obwohl unter Entwicklern Pytorch immer noch beliebter ist als Tensorflow. Aber in Zukunft werden möglicherweise mehr große Modelle auf Basis der JAX-Plattform trainiert und ausgeführt. Modelle Kürzlich hat das Keras-Team drei Backends (TensorFlow, JAX, PyTorch) mit der nativen PyTorch-Implementierung und Keras2 mit TensorFlow verglichen. Zunächst wählen sie eine Reihe von Mainstream-Inhalten aus
 Mar 18, 2024 am 09:20 AM
Mar 18, 2024 am 09:20 AM
Heute möchte ich eine aktuelle Forschungsarbeit der University of Connecticut vorstellen, die eine Methode zum Abgleichen von Zeitreihendaten mit großen NLP-Modellen (Natural Language Processing) im latenten Raum vorschlägt, um die Leistung von Zeitreihenprognosen zu verbessern. Der Schlüssel zu dieser Methode besteht darin, latente räumliche Hinweise (Eingabeaufforderungen) zu verwenden, um die Genauigkeit von Zeitreihenvorhersagen zu verbessern. Titel des Papiers: S2IP-LLM: SemanticSpaceInformedPromptLearningwithLLMforTimeSeriesForecasting Download-Adresse: https://arxiv.org/pdf/2403.05798v1.pdf 1. Hintergrundmodell für große Probleme
 Tesla-Roboter arbeiten in Fabriken, Musk: Der Freiheitsgrad der Hände wird dieses Jahr 22 erreichen!
May 06, 2024 pm 04:13 PM
Tesla-Roboter arbeiten in Fabriken, Musk: Der Freiheitsgrad der Hände wird dieses Jahr 22 erreichen!
May 06, 2024 pm 04:13 PM
Das neueste Video von Teslas Roboter Optimus ist veröffentlicht und er kann bereits in der Fabrik arbeiten. Bei normaler Geschwindigkeit sortiert es Batterien (Teslas 4680-Batterien) so: Der Beamte hat auch veröffentlicht, wie es bei 20-facher Geschwindigkeit aussieht – auf einer kleinen „Workstation“, pflücken und pflücken und pflücken: Dieses Mal wird es freigegeben. Eines der Highlights Der Vorteil des Videos besteht darin, dass Optimus diese Arbeit in der Fabrik völlig autonom und ohne menschliches Eingreifen während des gesamten Prozesses erledigt. Und aus Sicht von Optimus kann es auch die krumme Batterie aufnehmen und platzieren, wobei der Schwerpunkt auf der automatischen Fehlerkorrektur liegt: In Bezug auf die Hand von Optimus gab der NVIDIA-Wissenschaftler Jim Fan eine hohe Bewertung ab: Die Hand von Optimus ist der fünffingrige Roboter der Welt am geschicktesten. Seine Hände sind nicht nur taktil
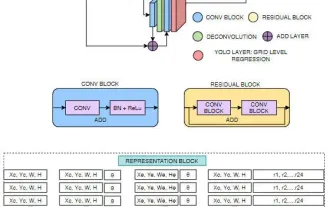 FisheyeDetNet: der erste Zielerkennungsalgorithmus basierend auf einer Fischaugenkamera
Apr 26, 2024 am 11:37 AM
FisheyeDetNet: der erste Zielerkennungsalgorithmus basierend auf einer Fischaugenkamera
Apr 26, 2024 am 11:37 AM
Die Zielerkennung ist ein relativ ausgereiftes Problem in autonomen Fahrsystemen, wobei die Fußgängererkennung einer der ersten Algorithmen ist, die eingesetzt werden. In den meisten Arbeiten wurde eine sehr umfassende Recherche durchgeführt. Die Entfernungswahrnehmung mithilfe von Fischaugenkameras für die Rundumsicht ist jedoch relativ wenig untersucht. Aufgrund der großen radialen Verzerrung ist es schwierig, die standardmäßige Bounding-Box-Darstellung in Fischaugenkameras zu implementieren. Um die obige Beschreibung zu vereinfachen, untersuchen wir erweiterte Begrenzungsrahmen-, Ellipsen- und allgemeine Polygondesigns in Polar-/Winkeldarstellungen und definieren eine mIOU-Metrik für die Instanzsegmentierung, um diese Darstellungen zu analysieren. Das vorgeschlagene Modell „fisheyeDetNet“ mit polygonaler Form übertrifft andere Modelle und erreicht gleichzeitig 49,5 % mAP auf dem Valeo-Fisheye-Kameradatensatz für autonomes Fahren
