
 Technologie-Peripheriegeräte
KI
Das MIT-Team nutzte eine autonome Molekülentdeckungsplattform mit geschlossenem Regelkreis für maschinelles Lernen, um 303 neue Moleküle erfolgreich zu entdecken, zu synthetisieren und zu beschreiben.
Technologie-Peripheriegeräte
KI
Das MIT-Team nutzte eine autonome Molekülentdeckungsplattform mit geschlossenem Regelkreis für maschinelles Lernen, um 303 neue Moleküle erfolgreich zu entdecken, zu synthetisieren und zu beschreiben.
Das MIT-Team nutzte eine autonome Molekülentdeckungsplattform mit geschlossenem Regelkreis für maschinelles Lernen, um 303 neue Moleküle erfolgreich zu entdecken, zu synthetisieren und zu beschreiben.


Herausgeber |
Da Chemiker zunehmend automatisierte Geräte und prädiktive Synthesealgorithmen verwenden, kommen autonome Forschungsgeräte der Realität immer näher.
Kürzlich haben Forscher des MIT eine autonome molekulare Entdeckungsplattform mit geschlossenem Regelkreis entwickelt, die durch integrierte Tools für maschinelles Lernen angetrieben wird, um das Design von Molekülen mit den gewünschten Eigenschaften zu beschleunigen. Erkunden Sie den chemischen Raum und nutzen Sie bekannte chemische Strukturen ohne manuelle Experimente.
In zwei Fallstudien versuchte die Plattform über 3000 Reaktionen, von denen über 1000 vorhergesagte Reaktionsprodukte erzeugten, und 303 nicht gemeldete farbstoffähnliche Moleküle wurden vorgeschlagen, synthetisiert und charakterisiert.
Die Studie mit dem Titel „Autonome, multiproperty-getriebene molekulare Entdeckung: Von Vorhersagen zu Messungen und zurück“ wurde am 22. Dezember 2023 in „Science“ veröffentlicht.
 Link zum Papier: https://www.science.org/doi/10.1126/science.adi1407
Link zum Papier: https://www.science.org/doi/10.1126/science.adi1407
Die Entdeckung kleiner Moleküle mit gewünschten funktionellen Eigenschaften ist entscheidend für Fortschritte in den Bereichen Gesundheit, Energie und nachhaltige Entwicklung. Der Prozess verläuft typischerweise durch einen langsamen, mühsamen, iterativen Design-Make-Test-Analyze (DMTA)-Zyklus.
Neue Tools für maschinelles Lernen (ML) können durch computergestützte Syntheseplanung (CASP) neue Kandidatenmoleküle generieren, ihre Eigenschaften vorhersagen und Reaktionswege vorschlagen. Fortschritte in der Automatisierung der Chemie ermöglichen die chemische Synthese und Charakterisierung mit minimalem menschlichen Eingriff nach manueller Einrichtung.
Die Integration von ML-Generierungsalgorithmen, ML-Eigenschaftsvorhersage, CASP, Robotik und automatisierter chemischer Synthese, Reinigung und Charakterisierung in DMTA-Workflows ermöglicht die Entwicklung autonomer chemischer Entdeckungsplattformen, die in verschiedenen chemischen Räumen betrieben werden können, ohne dass eine manuelle Neukonfiguration erforderlich ist. Die ideale eigenschaftsorientierte Entdeckungsplattform würde Moleküle vorschlagen und synthetisieren, um die Generierung und Eigenschaftsmodelle durch maschinelles Lernen zu bereichern und letztendlich die leistungsstärksten Moleküle zu entdecken. In der Praxis gilt es, Reaktionen auszuschließen, die von der verfügbaren Automatisierungshardware nicht sicher ausgeführt werden können.
Um eine autonome Entdeckung zu ermöglichen, demonstriert ein Forschungsteam des MIT eine integrierte DMTA-Schleife, die iterativ Moleküle vorschlägt, implementiert und charakterisiert und dabei den chemischen Raum nur mithilfe von Vorhersagetools erforscht.
Graph-Completion generiert Modelle zum Entwurf von Kandidatenmolekülen und verwendet ML-Modelle, um sie für jede dieser drei Eigenschaften zu bewerten. Das CASP-Tool schlägt mehrstufige Syntheserezepte vor, die von automatisierten Liquid-Handlern, Batch-Reaktoren, Hochleistungsflüssigkeitschromatographie (HPLC) und Roboterarmen durchgeführt werden. Der Plattenleser misst Absorptionsspektren, kalibrierte HPLC-Retentionszeiten liefern Wasser-Oktanol-Verteilungskoeffizienten und eine simulierte Sonnenlichtquelle wird mit dem Plattenleser kombiniert, um den photooxidativen Abbau zu quantifizieren. Gemessene molekulare Eigenschaften werden automatisch zurückgemeldet, um das Eigenschaftsvorhersagemodell neu zu trainieren und so einen Schritt des automatisierten DMTA-Zyklus abzuschließen.
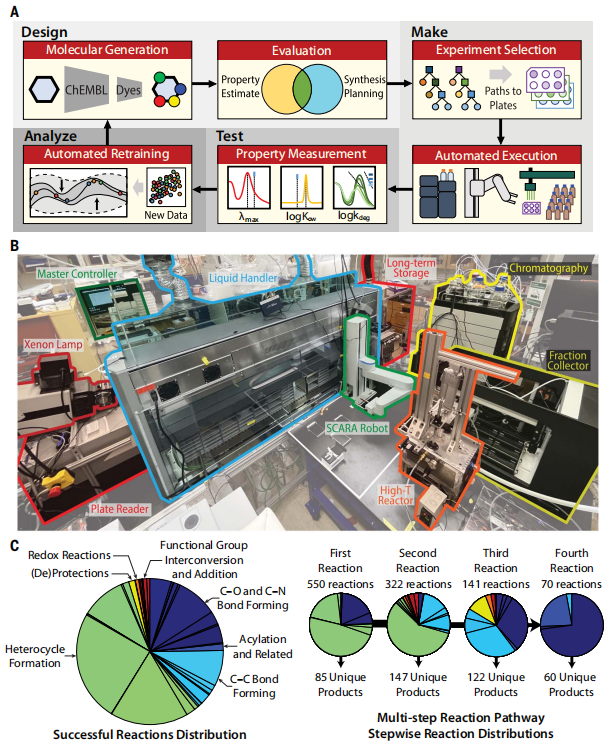 Integrierte Plattformübersicht und von der Plattform vorhergesagte und erfolgreich ausgeführte Reaktionen. (Quelle: Papier)
Integrierte Plattformübersicht und von der Plattform vorhergesagte und erfolgreich ausgeführte Reaktionen. (Quelle: Papier)
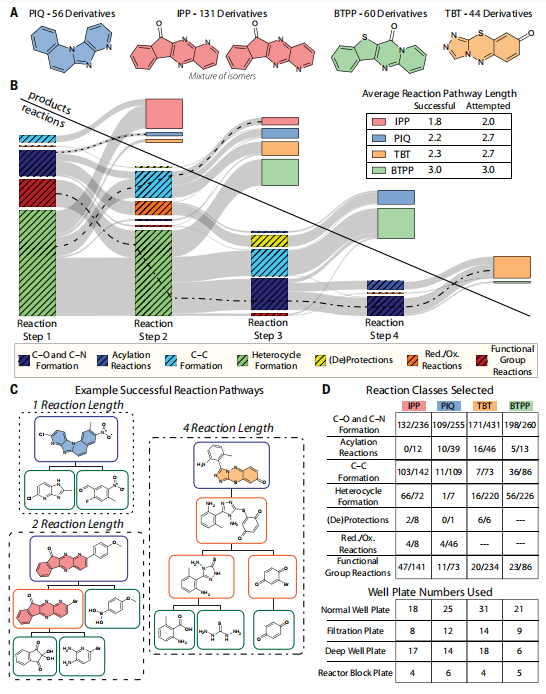 Erkunden Sie chemische Vorhersagen und Versuche anhand von Fallstudien. (Quelle: Paper)
Erkunden Sie chemische Vorhersagen und Versuche anhand von Fallstudien. (Quelle: Paper)
Für beide Fallstudien waren Absorptionsmaximum, Verteilungskoeffizient und Photooxidationsstabilität die Zieleigenschaften, und die Plattform hat jede Eigenschaft automatisch gemessen und aufgezeichnet, um Modellvorhersagen zu verfeinern und zukünftige experimentelle Entscheidungen zu treffen.
Die Forscher sagten: „Zukünftige Iterationen der Plattform werden von Verbesserungen der Vorhersagefähigkeiten profitieren, insbesondere der Reaktionstreue, der Zustandsempfehlung und der Molekülgenerierung sowie der Analysetools. Die kontinuierliche Entwicklung der integrierten Plattform mit geschlossenem Regelkreis ist eine Möglichkeit, weiterzumachen.“ Beschleunigung der molekularen Entdeckung. „Der Weg der Hoffnung.“
Das obige ist der detaillierte Inhalt vonDas MIT-Team nutzte eine autonome Molekülentdeckungsplattform mit geschlossenem Regelkreis für maschinelles Lernen, um 303 neue Moleküle erfolgreich zu entdecken, zu synthetisieren und zu beschreiben.. Für weitere Informationen folgen Sie bitte anderen verwandten Artikeln auf der PHP chinesischen Website!

Heiße KI -Werkzeuge

Undresser.AI Undress
KI-gestützte App zum Erstellen realistischer Aktfotos

AI Clothes Remover
Online-KI-Tool zum Entfernen von Kleidung aus Fotos.

Undress AI Tool
Ausziehbilder kostenlos

Clothoff.io
KI-Kleiderentferner

AI Hentai Generator
Erstellen Sie kostenlos Ai Hentai.

Heißer Artikel
Heiße Werkzeuge

Notepad++7.3.1
Einfach zu bedienender und kostenloser Code-Editor

SublimeText3 chinesische Version
Chinesische Version, sehr einfach zu bedienen

Senden Sie Studio 13.0.1
Leistungsstarke integrierte PHP-Entwicklungsumgebung

Dreamweaver CS6
Visuelle Webentwicklungstools

SublimeText3 Mac-Version
Codebearbeitungssoftware auf Gottesniveau (SublimeText3)
Heiße Themen
 1376
1376
 52
52

 „Defect Spectrum' durchbricht die Grenzen der herkömmlichen Fehlererkennung und erreicht erstmals eine hochpräzise und umfassende semantische Fehlererkennung in der Industrie.
Jul 26, 2024 pm 05:38 PM
„Defect Spectrum' durchbricht die Grenzen der herkömmlichen Fehlererkennung und erreicht erstmals eine hochpräzise und umfassende semantische Fehlererkennung in der Industrie.
Jul 26, 2024 pm 05:38 PM
In der modernen Fertigung ist die genaue Fehlererkennung nicht nur der Schlüssel zur Sicherstellung der Produktqualität, sondern auch der Kern für die Verbesserung der Produktionseffizienz. Allerdings mangelt es vorhandenen Datensätzen zur Fehlererkennung häufig an der Genauigkeit und dem semantischen Reichtum, die für praktische Anwendungen erforderlich sind, was dazu führt, dass Modelle bestimmte Fehlerkategorien oder -orte nicht identifizieren können. Um dieses Problem zu lösen, hat ein Spitzenforschungsteam bestehend aus der Hong Kong University of Science and Technology Guangzhou und Simou Technology innovativ den „DefectSpectrum“-Datensatz entwickelt, der eine detaillierte und semantisch reichhaltige groß angelegte Annotation von Industriedefekten ermöglicht. Wie in Tabelle 1 gezeigt, bietet der Datensatz „DefectSpectrum“ im Vergleich zu anderen Industriedatensätzen die meisten Fehleranmerkungen (5438 Fehlerproben) und die detaillierteste Fehlerklassifizierung (125 Fehlerkategorien).
 Das NVIDIA-Dialogmodell ChatQA wurde auf Version 2.0 weiterentwickelt, wobei die angegebene Kontextlänge 128 KB beträgt
Jul 26, 2024 am 08:40 AM
Das NVIDIA-Dialogmodell ChatQA wurde auf Version 2.0 weiterentwickelt, wobei die angegebene Kontextlänge 128 KB beträgt
Jul 26, 2024 am 08:40 AM
Die offene LLM-Community ist eine Ära, in der hundert Blumen blühen und konkurrieren. Sie können Llama-3-70B-Instruct, QWen2-72B-Instruct, Nemotron-4-340B-Instruct, Mixtral-8x22BInstruct-v0.1 und viele andere sehen hervorragende Darsteller. Allerdings weisen offene Modelle im Vergleich zu den proprietären Großmodellen GPT-4-Turbo in vielen Bereichen noch erhebliche Lücken auf. Zusätzlich zu allgemeinen Modellen wurden einige offene Modelle entwickelt, die sich auf Schlüsselbereiche spezialisieren, wie etwa DeepSeek-Coder-V2 für Programmierung und Mathematik und InternVL für visuelle Sprachaufgaben.
 Google AI gewann die Silbermedaille der IMO Mathematical Olympiad, das mathematische Argumentationsmodell AlphaProof wurde eingeführt und Reinforcement Learning ist zurück
Jul 26, 2024 pm 02:40 PM
Google AI gewann die Silbermedaille der IMO Mathematical Olympiad, das mathematische Argumentationsmodell AlphaProof wurde eingeführt und Reinforcement Learning ist zurück
Jul 26, 2024 pm 02:40 PM
Für KI ist die Mathematikolympiade kein Problem mehr. Am Donnerstag hat die künstliche Intelligenz von Google DeepMind eine Meisterleistung vollbracht: Sie nutzte KI, um meiner Meinung nach die eigentliche Frage der diesjährigen Internationalen Mathematikolympiade zu lösen, und war nur einen Schritt davon entfernt, die Goldmedaille zu gewinnen. Der IMO-Wettbewerb, der gerade letzte Woche zu Ende ging, hatte sechs Fragen zu Algebra, Kombinatorik, Geometrie und Zahlentheorie. Das von Google vorgeschlagene hybride KI-System beantwortete vier Fragen richtig und erzielte 28 Punkte und erreichte damit die Silbermedaillenstufe. Anfang dieses Monats hatte der UCLA-Professor Terence Tao gerade die KI-Mathematische Olympiade (AIMO Progress Award) mit einem Millionenpreis gefördert. Unerwarteterweise hatte sich das Niveau der KI-Problemlösung vor Juli auf dieses Niveau verbessert. Beantworten Sie die Fragen meiner Meinung nach gleichzeitig. Am schwierigsten ist es meiner Meinung nach, da sie die längste Geschichte, den größten Umfang und die negativsten Fragen haben
 Der Standpunkt der Natur: Die Erprobung künstlicher Intelligenz in der Medizin ist im Chaos. Was ist zu tun?
Aug 22, 2024 pm 04:37 PM
Der Standpunkt der Natur: Die Erprobung künstlicher Intelligenz in der Medizin ist im Chaos. Was ist zu tun?
Aug 22, 2024 pm 04:37 PM
Herausgeber | ScienceAI Basierend auf begrenzten klinischen Daten wurden Hunderte medizinischer Algorithmen genehmigt. Wissenschaftler diskutieren darüber, wer die Werkzeuge testen soll und wie dies am besten geschieht. Devin Singh wurde Zeuge, wie ein pädiatrischer Patient in der Notaufnahme einen Herzstillstand erlitt, während er lange auf eine Behandlung wartete, was ihn dazu veranlasste, den Einsatz von KI zu erforschen, um Wartezeiten zu verkürzen. Mithilfe von Triage-Daten aus den Notaufnahmen von SickKids erstellten Singh und Kollegen eine Reihe von KI-Modellen, um mögliche Diagnosen zu stellen und Tests zu empfehlen. Eine Studie zeigte, dass diese Modelle die Zahl der Arztbesuche um 22,3 % verkürzen können und die Verarbeitung der Ergebnisse pro Patient, der einen medizinischen Test benötigt, um fast drei Stunden beschleunigt. Der Erfolg von Algorithmen der künstlichen Intelligenz in der Forschung bestätigt dies jedoch nur
 Training mit Millionen von Kristalldaten zur Lösung kristallographischer Phasenprobleme, die Deep-Learning-Methode PhAI wird in Science veröffentlicht
Aug 08, 2024 pm 09:22 PM
Training mit Millionen von Kristalldaten zur Lösung kristallographischer Phasenprobleme, die Deep-Learning-Methode PhAI wird in Science veröffentlicht
Aug 08, 2024 pm 09:22 PM
Herausgeber |KX Bis heute sind die durch die Kristallographie ermittelten Strukturdetails und Präzision, von einfachen Metallen bis hin zu großen Membranproteinen, mit keiner anderen Methode zu erreichen. Die größte Herausforderung, das sogenannte Phasenproblem, bleibt jedoch die Gewinnung von Phaseninformationen aus experimentell bestimmten Amplituden. Forscher der Universität Kopenhagen in Dänemark haben eine Deep-Learning-Methode namens PhAI entwickelt, um Kristallphasenprobleme zu lösen. Ein Deep-Learning-Neuronales Netzwerk, das mithilfe von Millionen künstlicher Kristallstrukturen und den entsprechenden synthetischen Beugungsdaten trainiert wird, kann genaue Elektronendichtekarten erstellen. Die Studie zeigt, dass diese Deep-Learning-basierte Ab-initio-Strukturlösungsmethode das Phasenproblem mit einer Auflösung von nur 2 Angström lösen kann, was nur 10 bis 20 % der bei atomarer Auflösung verfügbaren Daten im Vergleich zur herkömmlichen Ab-initio-Berechnung entspricht
 Um ein neues wissenschaftliches und komplexes Frage-Antwort-Benchmark- und Bewertungssystem für große Modelle bereitzustellen, haben UNSW, Argonne, die University of Chicago und andere Institutionen gemeinsam das SciQAG-Framework eingeführt
Jul 25, 2024 am 06:42 AM
Um ein neues wissenschaftliches und komplexes Frage-Antwort-Benchmark- und Bewertungssystem für große Modelle bereitzustellen, haben UNSW, Argonne, die University of Chicago und andere Institutionen gemeinsam das SciQAG-Framework eingeführt
Jul 25, 2024 am 06:42 AM
Herausgeber | Der Frage-Antwort-Datensatz (QA) von ScienceAI spielt eine entscheidende Rolle bei der Förderung der Forschung zur Verarbeitung natürlicher Sprache (NLP). Hochwertige QS-Datensätze können nicht nur zur Feinabstimmung von Modellen verwendet werden, sondern auch effektiv die Fähigkeiten großer Sprachmodelle (LLMs) bewerten, insbesondere die Fähigkeit, wissenschaftliche Erkenntnisse zu verstehen und zu begründen. Obwohl es derzeit viele wissenschaftliche QS-Datensätze aus den Bereichen Medizin, Chemie, Biologie und anderen Bereichen gibt, weisen diese Datensätze immer noch einige Mängel auf. Erstens ist das Datenformular relativ einfach, die meisten davon sind Multiple-Choice-Fragen. Sie sind leicht auszuwerten, schränken jedoch den Antwortauswahlbereich des Modells ein und können die Fähigkeit des Modells zur Beantwortung wissenschaftlicher Fragen nicht vollständig testen. Im Gegensatz dazu offene Fragen und Antworten
 Identifizieren Sie automatisch die besten Moleküle und reduzieren Sie die Synthesekosten. Das MIT entwickelt ein Algorithmus-Framework für die Entscheidungsfindung im molekularen Design
Jun 22, 2024 am 06:43 AM
Identifizieren Sie automatisch die besten Moleküle und reduzieren Sie die Synthesekosten. Das MIT entwickelt ein Algorithmus-Framework für die Entscheidungsfindung im molekularen Design
Jun 22, 2024 am 06:43 AM
Herausgeber |. Der Einsatz von Ziluo AI bei der Rationalisierung der Arzneimittelforschung nimmt explosionsartig zu. Durchsuchen Sie Milliarden von Kandidatenmolekülen nach solchen, die möglicherweise über Eigenschaften verfügen, die für die Entwicklung neuer Medikamente erforderlich sind. Es sind so viele Variablen zu berücksichtigen, von Materialpreisen bis hin zum Fehlerrisiko, dass es keine leichte Aufgabe ist, die Kosten für die Synthese der besten Kandidatenmoleküle abzuwägen, selbst wenn Wissenschaftler KI einsetzen. Hier entwickelten MIT-Forscher SPARROW, ein quantitatives Entscheidungsalgorithmus-Framework, um automatisch die besten molekularen Kandidaten zu identifizieren und so die Synthesekosten zu minimieren und gleichzeitig die Wahrscheinlichkeit zu maximieren, dass die Kandidaten die gewünschten Eigenschaften aufweisen. Der Algorithmus bestimmte auch die Materialien und experimentellen Schritte, die zur Synthese dieser Moleküle erforderlich sind. SPARROW berücksichtigt die Kosten für die gleichzeitige Synthese einer Charge von Molekülen, da häufig mehrere Kandidatenmoleküle verfügbar sind
 SOTA Performance, eine multimodale KI-Methode zur Vorhersage der Protein-Ligand-Affinität in Xiamen, kombiniert erstmals molekulare Oberflächeninformationen
Jul 17, 2024 pm 06:37 PM
SOTA Performance, eine multimodale KI-Methode zur Vorhersage der Protein-Ligand-Affinität in Xiamen, kombiniert erstmals molekulare Oberflächeninformationen
Jul 17, 2024 pm 06:37 PM
Herausgeber |. KX Im Bereich der Arzneimittelforschung und -entwicklung ist die genaue und effektive Vorhersage der Bindungsaffinität von Proteinen und Liganden für das Arzneimittelscreening und die Arzneimitteloptimierung von entscheidender Bedeutung. Aktuelle Studien berücksichtigen jedoch nicht die wichtige Rolle molekularer Oberflächeninformationen bei Protein-Ligand-Wechselwirkungen. Auf dieser Grundlage schlugen Forscher der Universität Xiamen ein neuartiges Framework zur multimodalen Merkmalsextraktion (MFE) vor, das erstmals Informationen über Proteinoberfläche, 3D-Struktur und -Sequenz kombiniert und einen Kreuzaufmerksamkeitsmechanismus verwendet, um verschiedene Modalitäten zu vergleichen Ausrichtung. Experimentelle Ergebnisse zeigen, dass diese Methode bei der Vorhersage von Protein-Ligand-Bindungsaffinitäten Spitzenleistungen erbringt. Darüber hinaus belegen Ablationsstudien die Wirksamkeit und Notwendigkeit der Proteinoberflächeninformation und der multimodalen Merkmalsausrichtung innerhalb dieses Rahmens. Verwandte Forschungen beginnen mit „S
