
 Technologie-Peripheriegeräte
KI
KI wird nicht gelernt! Neue Forschungsergebnisse zeigen Wege auf, die Blackbox der künstlichen Intelligenz zu entschlüsseln
Technologie-Peripheriegeräte
KI
KI wird nicht gelernt! Neue Forschungsergebnisse zeigen Wege auf, die Blackbox der künstlichen Intelligenz zu entschlüsseln
KI wird nicht gelernt! Neue Forschungsergebnisse zeigen Wege auf, die Blackbox der künstlichen Intelligenz zu entschlüsseln

Künstliche Intelligenz (KI) hat sich rasant weiterentwickelt, doch für den Menschen sind leistungsstarke Modelle eine „Black Box“.
Wir verstehen das Innenleben des Modells und den Prozess, durch den es zu seinen Schlussfolgerungen gelangt, nicht.
Doch kürzlich gelang Professor Jürgen Bajorath, Cheminformatiker an der Universität Bonn, und seinem Team ein großer Durchbruch.
Sie haben eine Technik entwickelt, die zeigt, wie einige Systeme der künstlichen Intelligenz funktionieren, die in der Arzneimittelforschung eingesetzt werden.
Untersuchungen zeigen, dass Modelle der künstlichen Intelligenz die Wirksamkeit von Medikamenten hauptsächlich durch den Abruf vorhandener Daten vorhersagen, anstatt spezifische chemische Wechselwirkungen zu lernen.
—Mit anderen Worten, KI-Vorhersagen basieren ausschließlich auf dem Zusammensetzen von Erinnerungen, und maschinelles Lernen lernt nicht wirklich!
Ihre Forschungsergebnisse wurden kürzlich in der Zeitschrift Nature Machine Intelligence veröffentlicht.

Papieradresse: https://www.nature.com/articles/s42256-023-00756-9
In der Medizin suchen Forscher fieberhaft nach wirksamen Wirkstoffen zur Bekämpfung von Krankheiten —Welche Wirkstoffmoleküle sind am wirksamsten?
Üblicherweise werden diese wirksamen Moleküle (Verbindungen) an Proteine angedockt, die als Enzyme oder Rezeptoren fungieren und bestimmte physiologische Wirkungsketten auslösen.
In besonderen Fällen sind bestimmte Moleküle auch dafür verantwortlich, unerwünschte Reaktionen im Körper, wie zum Beispiel überschießende Entzündungsreaktionen, zu blockieren.
Die Anzahl der möglichen Verbindungen ist riesig und diejenige zu finden, die funktioniert, ist wie die Suche nach der Nadel im Heuhaufen.
Also nutzten die Forscher zunächst KI-Modelle, um vorherzusagen, welche Moleküle am besten andocken und sich stark an ihre jeweiligen Zielproteine binden würden. Diese Medikamentenkandidaten werden dann in experimentellen Studien noch detaillierter untersucht.
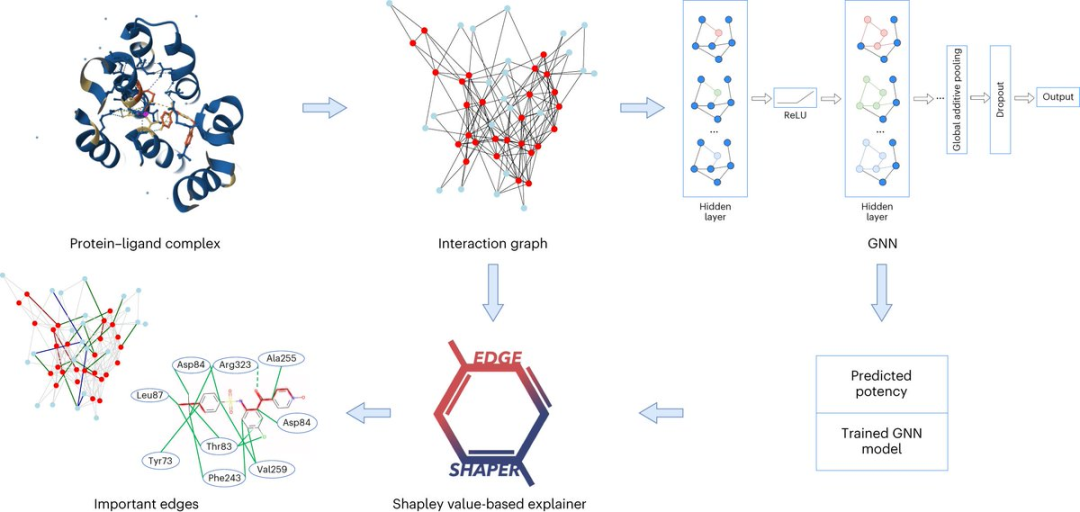
Seit der Entwicklung der künstlichen Intelligenz werden in der Arzneimittelforschung zunehmend KI-bezogene Technologien eingesetzt.
Graph Neural Network (GNN) eignet sich beispielsweise zur Vorhersage der Bindungsstärke eines bestimmten Moleküls an ein Zielprotein.
Ein Diagramm besteht aus Knoten, die Objekte darstellen, und Kanten, die Beziehungen zwischen Knoten darstellen. In der Diagrammdarstellung eines Protein-Ligand-Komplexes verbinden die Kanten des Diagramms Protein- oder Ligandenknoten und stellen die Struktur einer Substanz oder die Wechselwirkung zwischen einem Protein und einem Liganden dar.
Das GNN-Modell verwendet aus Röntgenstrukturen extrahierte Protein-Ligand-Interaktionskarten, um Ligandenaffinitäten vorherzusagen.
Professor Jürgen Bajorath sagte, dass das GNN-Modell für uns wie eine Blackbox ist und wir keine Möglichkeit haben zu wissen, wie es seine Vorhersagen ableitet.

Professor Jürgen Bajorath arbeitet am LIMES-Institut der Universität Bonn, am Bonn-Aachen International Center for Information Technology (Bonn-Aachen International Center for Information Technology) und am Lamarr Institute for Machine Learning and Artificial Intelligence (Lamarr-Institut für maschinelles Lernen und künstliche Intelligenz).
Wie funktioniert künstliche Intelligenz?
Forscher der Abteilung für Chemische Informatik der Universität Bonn haben gemeinsam mit Kollegen der Sapienza-Universität Rom detailliert analysiert, ob das graphische neuronale Netzwerk die Interaktion zwischen dem Protein und dem Liganden wirklich gelernt hat.
Die Forscher analysierten insgesamt sechs verschiedene GNN-Architekturen mit ihrer speziell entwickelten Methode „EdgeSHAPer“.
Das EdgeSHAPer-Programm kann feststellen, ob das GNN die wichtigsten Wechselwirkungen zwischen Verbindungen und Proteinen gelernt oder mit anderen Methoden Vorhersagen getroffen hat.
Die Wissenschaftler trainierten sechs GNNs mithilfe von Diagrammen, die aus den Strukturen von Protein-Ligand-Komplexen extrahiert wurden – wobei die Wirkungsweise der Verbindung und die Stärke ihrer Bindung an das Zielprotein bekannt waren.
Testen Sie dann das trainierte GNN an anderen Verbindungen und verwenden Sie EdgeSHAPer, um zu analysieren, wie das GNN Vorhersagen erzeugt.
„Wenn sich GNNs wie erwartet verhalten, müssen sie die Wechselwirkungen zwischen Verbindungen und Zielproteinen lernen und Vorhersagen treffen, indem sie bestimmte Wechselwirkungen priorisieren.“
Der Analyse des Forschungsteams zufolge gelang dies jedoch sechs GNNs grundsätzlich nicht. Die meisten GNNs lernen nur einige Protein-Arzneimittel-Wechselwirkungen und konzentrieren sich hauptsächlich auf Liganden.
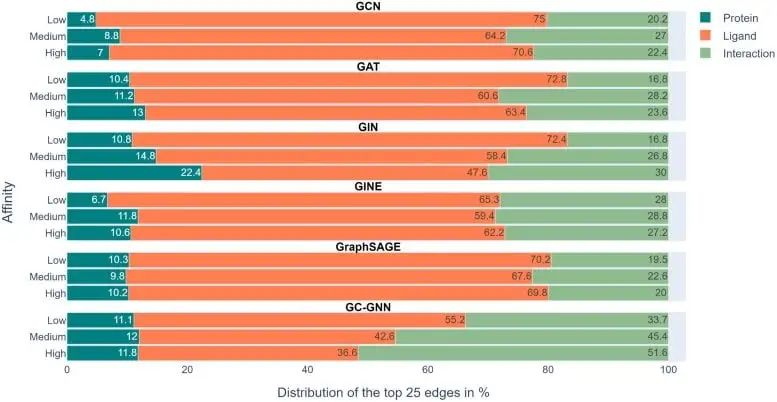
Die obige Abbildung zeigt die experimentellen Ergebnisse in 6 GNNs. Der Farbskalenbalken stellt den durchschnittlichen Anteil von Proteinen, Liganden und Wechselwirkungen in den oberen 25 Kanten jeder von EdgeSHAPer ermittelten Vorhersage dar.
Wir können sehen, dass das Modell die durch Grün dargestellte Interaktion lernen muss, ihr Anteil im gesamten Experiment jedoch nicht hoch ist und der orangefarbene Balken, der den Liganden darstellt, den größten Anteil ausmacht.
Um die Bindungsstärke eines Moleküls an ein Zielprotein vorherzusagen, „merken“ sich Modelle in erster Linie die chemisch ähnlichen Moleküle und ihre Bindungsdaten, auf die sie während des Trainings gestoßen sind, unabhängig vom Zielprotein. Diese erinnerten chemischen Ähnlichkeiten bestimmen im Wesentlichen die Vorhersage.
Das erinnert an den „Clever-Hans-Effekt“ – genau wie das Pferd, das scheinbar zählen kann, sich aber tatsächlich auf die Nuancen der Mimik und Gestik seiner Gefährten stützt, um auf das Erwartete zu schließen Ergebnisse.
Dies kann bedeuten, dass die sogenannte „Lernfähigkeit“ von GNN möglicherweise unhaltbar ist und die Vorhersagen des Modells weitgehend überschätzt werden, da chemische Kenntnisse und einfachere Methoden verwendet werden können, um Vorhersagen mit derselben Qualität durchzuführen.
Allerdings wurde in der Studie auch ein weiteres Phänomen entdeckt: Wenn die Wirksamkeit der Testverbindung zunimmt, neigt das Modell dazu, mehr Interaktionen zu lernen.
Vielleicht können diese GNNs durch Modifizierung der Darstellungs- und Trainingstechniken weiter in die gewünschte Richtung verbessert werden. Allerdings ist die Annahme, dass physikalische Größen aus molekularen Graphen gelernt werden können, grundsätzlich mit Vorsicht zu genießen.
„Künstliche Intelligenz ist keine schwarze Magie.“
Das obige ist der detaillierte Inhalt vonKI wird nicht gelernt! Neue Forschungsergebnisse zeigen Wege auf, die Blackbox der künstlichen Intelligenz zu entschlüsseln. Für weitere Informationen folgen Sie bitte anderen verwandten Artikeln auf der PHP chinesischen Website!

Heiße KI -Werkzeuge

Undresser.AI Undress
KI-gestützte App zum Erstellen realistischer Aktfotos

AI Clothes Remover
Online-KI-Tool zum Entfernen von Kleidung aus Fotos.

Undress AI Tool
Ausziehbilder kostenlos

Clothoff.io
KI-Kleiderentferner

AI Hentai Generator
Erstellen Sie kostenlos Ai Hentai.

Heißer Artikel
Heiße Werkzeuge

Notepad++7.3.1
Einfach zu bedienender und kostenloser Code-Editor

SublimeText3 chinesische Version
Chinesische Version, sehr einfach zu bedienen

Senden Sie Studio 13.0.1
Leistungsstarke integrierte PHP-Entwicklungsumgebung

Dreamweaver CS6
Visuelle Webentwicklungstools

SublimeText3 Mac-Version
Codebearbeitungssoftware auf Gottesniveau (SublimeText3)
Heiße Themen
 1378
1378
 52
52

 So optimieren Sie die Leistung von Debian Readdir
Apr 13, 2025 am 08:48 AM
So optimieren Sie die Leistung von Debian Readdir
Apr 13, 2025 am 08:48 AM
In Debian -Systemen werden Readdir -Systemaufrufe zum Lesen des Verzeichnisinhalts verwendet. Wenn seine Leistung nicht gut ist, probieren Sie die folgende Optimierungsstrategie aus: Vereinfachen Sie die Anzahl der Verzeichnisdateien: Teilen Sie große Verzeichnisse so weit wie möglich in mehrere kleine Verzeichnisse auf und reduzieren Sie die Anzahl der gemäß Readdir -Anrufe verarbeiteten Elemente. Aktivieren Sie den Verzeichnis -Inhalt Caching: Erstellen Sie einen Cache -Mechanismus, aktualisieren Sie den Cache regelmäßig oder bei Änderungen des Verzeichnisinhalts und reduzieren Sie häufige Aufrufe an Readdir. Speicher -Caches (wie Memcached oder Redis) oder lokale Caches (wie Dateien oder Datenbanken) können berücksichtigt werden. Nehmen Sie eine effiziente Datenstruktur an: Wenn Sie das Verzeichnis -Traversal selbst implementieren, wählen Sie effizientere Datenstrukturen (z.
 So implementieren Sie die Dateisortierung nach Debian Readdir
Apr 13, 2025 am 09:06 AM
So implementieren Sie die Dateisortierung nach Debian Readdir
Apr 13, 2025 am 09:06 AM
In Debian -Systemen wird die Readdir -Funktion zum Lesen des Verzeichnisinhalts verwendet, aber die Reihenfolge, in der sie zurückgibt, ist nicht vordefiniert. Um Dateien in einem Verzeichnis zu sortieren, müssen Sie zuerst alle Dateien lesen und dann mit der QSORT -Funktion sortieren. Der folgende Code zeigt, wie Verzeichnisdateien mithilfe von Readdir und QSORT in Debian System sortiert werden:#include#include#include#include // benutzerdefinierte Vergleichsfunktion, verwendet für QSortIntCompare (constvoid*a, constvoid*b) {rettrcmp (*(*(*(
 So setzen Sie die Debian Apache -Protokollebene fest
Apr 13, 2025 am 08:33 AM
So setzen Sie die Debian Apache -Protokollebene fest
Apr 13, 2025 am 08:33 AM
In diesem Artikel wird beschrieben, wie Sie die Protokollierungsstufe des Apacheweb -Servers im Debian -System anpassen. Durch Ändern der Konfigurationsdatei können Sie die ausführliche Ebene der von Apache aufgezeichneten Protokollinformationen steuern. Methode 1: Ändern Sie die Hauptkonfigurationsdatei, um die Konfigurationsdatei zu finden: Die Konfigurationsdatei von Apache2.x befindet sich normalerweise im Verzeichnis/etc/apache2/. Der Dateiname kann je nach Installationsmethode Apache2.conf oder httpd.conf sein. Konfigurationsdatei bearbeiten: Öffnen Sie die Konfigurationsdatei mit Stammberechtigungen mit einem Texteditor (z. B. Nano): Sudonano/etc/apache2/apache2.conf
 Wie Debian OpenSSL verhindert, dass Mann-in-the-Middle-Angriffe
Apr 13, 2025 am 10:30 AM
Wie Debian OpenSSL verhindert, dass Mann-in-the-Middle-Angriffe
Apr 13, 2025 am 10:30 AM
In Debian Systems ist OpenSSL eine wichtige Bibliothek für Verschlüsselung, Entschlüsselung und Zertifikatverwaltung. Um einen Mann-in-the-Middle-Angriff (MITM) zu verhindern, können folgende Maßnahmen ergriffen werden: Verwenden Sie HTTPS: Stellen Sie sicher, dass alle Netzwerkanforderungen das HTTPS-Protokoll anstelle von HTTP verwenden. HTTPS verwendet TLS (Transport Layer Security Protocol), um Kommunikationsdaten zu verschlüsseln, um sicherzustellen, dass die Daten während der Übertragung nicht gestohlen oder manipuliert werden. Überprüfen Sie das Serverzertifikat: Überprüfen Sie das Serverzertifikat im Client manuell, um sicherzustellen, dass es vertrauenswürdig ist. Der Server kann manuell durch die Delegate -Methode der URLSession überprüft werden
 Debian Mail Server SSL -Zertifikat -Installationsmethode
Apr 13, 2025 am 11:39 AM
Debian Mail Server SSL -Zertifikat -Installationsmethode
Apr 13, 2025 am 11:39 AM
Die Schritte zur Installation eines SSL -Zertifikats auf dem Debian Mail -Server sind wie folgt: 1. Installieren Sie zuerst das OpenSSL -Toolkit und stellen Sie sicher, dass das OpenSSL -Toolkit bereits in Ihrem System installiert ist. Wenn nicht installiert, können Sie den folgenden Befehl installieren: sudoapt-getupdatesudoapt-getinstallopenssl2. Generieren Sie den privaten Schlüssel und die Zertifikatanforderung als nächst
 Wie Debian Readdir sich in andere Tools integriert
Apr 13, 2025 am 09:42 AM
Wie Debian Readdir sich in andere Tools integriert
Apr 13, 2025 am 09:42 AM
Die Readdir -Funktion im Debian -System ist ein Systemaufruf, der zum Lesen des Verzeichnisgehalts verwendet wird und häufig in der C -Programmierung verwendet wird. In diesem Artikel wird erläutert, wie Readdir in andere Tools integriert wird, um seine Funktionalität zu verbessern. Methode 1: Kombinieren Sie C -Sprachprogramm und Pipeline zuerst ein C -Programm, um die Funktion der Readdir aufzurufen und das Ergebnis auszugeben:#include#include#includeIntmain (intargc, char*argv []) {Dir*Dir; structDirent*Eintrag; if (argc! = 2) {{
 Debian Mail Server Firewall -Konfigurationstipps
Apr 13, 2025 am 11:42 AM
Debian Mail Server Firewall -Konfigurationstipps
Apr 13, 2025 am 11:42 AM
Das Konfigurieren der Firewall eines Debian -Mailservers ist ein wichtiger Schritt zur Gewährleistung der Serversicherheit. Im Folgenden sind mehrere häufig verwendete Firewall -Konfigurationsmethoden, einschließlich der Verwendung von Iptables und Firewalld. Verwenden Sie Iptables, um Firewall so zu konfigurieren, dass Iptables (falls bereits installiert) installiert werden:
 Wie man Debian Syslog lernt
Apr 13, 2025 am 11:51 AM
Wie man Debian Syslog lernt
Apr 13, 2025 am 11:51 AM
In diesem Leitfaden werden Sie erfahren, wie Sie Syslog in Debian -Systemen verwenden. Syslog ist ein Schlüsseldienst in Linux -Systemen für Protokollierungssysteme und Anwendungsprotokollnachrichten. Es hilft den Administratoren, die Systemaktivitäten zu überwachen und zu analysieren, um Probleme schnell zu identifizieren und zu lösen. 1. Grundkenntnisse über syslog Die Kernfunktionen von Syslog umfassen: zentrales Sammeln und Verwalten von Protokollnachrichten; Unterstützung mehrerer Protokoll -Ausgabesformate und Zielorte (z. B. Dateien oder Netzwerke); Bereitstellung von Echtzeit-Protokoll- und Filterfunktionen. 2. Installieren und Konfigurieren von Syslog (mit Rsyslog) Das Debian -System verwendet standardmäßig Rsyslog. Sie können es mit dem folgenden Befehl installieren: sudoaptupdatesud
