


Saccharides are the most abundant organic substances in nature and are essential for life. Understanding how carbohydrates regulate proteins during physiological and pathological processes can provide opportunities to address key biological questions and develop new treatments.
However, the diversity and complexity of sugar molecules poses a challenge to experimentally identify sugar-protein binding and interaction sites.
Here, a team from the Chinese Academy of Sciences developed DeepGlycanSite, a deep learning model that is able to accurately predict sugar-binding sites on a given protein structure.
DeepGlycanSite integrates the geometric and evolutionary characteristics of proteins into a deep equivariant graph neural network with a Transformer architecture. Its performance significantly surpasses previous advanced methods and can effectively predict the binding sites of various sugar molecules.
Combined with mutagenesis studies, DeepGlycanSite reveals the guanosine-5'-bisphosphate recognition site of important G protein-coupled receptors.
These findings demonstrate the value of DeepGlycanSite for sugar binding site prediction and can provide insights into the molecular mechanisms behind sugar regulation of proteins of therapeutic importance.
The study was titled "Highly accurate carbohydrate-binding site prediction with DeepGlycanSite" and was published in "Nature Communications" on June 17, 2024.

Sugar is ubiquitous on the cell surface of all organisms. They interact with a variety of protein families such as lectins, antibodies, enzymes and transporters to regulate key biological processes such as immune response, cell differentiation and neural development. Understanding the interaction mechanism between carbohydrates and proteins is the basis for developing carbohydrate drugs.
However, the diversity and complexity of carbohydrate structures, especially the variability of their binding sites with proteins, pose challenges to the acquisition of experimental data and drug design. Illustration: The complexity of sugar molecules and the diversity of sugar binding sites. (Source: paper)
 In the past, traditional binding site prediction methods were not suitable for sugar molecules with complex structures and large changes in size. This, coupled with the scarcity of high-resolution structural data on sugar-protein complexes, limits the performance of predictive models.
In the past, traditional binding site prediction methods were not suitable for sugar molecules with complex structures and large changes in size. This, coupled with the scarcity of high-resolution structural data on sugar-protein complexes, limits the performance of predictive models.
In recent years, with the rapid development of Protein Data Bank (PDB) and open glycomics resources, the academic community has accumulated structural data of more than 19,000 such complexes. The increase in these high-quality data makes it possible to use AI technology to develop accurate sugar-binding site prediction models, which is expected to accelerate the discovery and optimization process of sugar drugs. In the latest research, the Chinese Academy of Sciences team introduced DeepGlycanSite, a deep equivariant graph neural network (EGNN) model that can accurately predict sugar-binding sites with target protein structures.
Illustration: DeepGlycanSite overview. (Source: paper)
 The team leveraged geometric features, such as directions and distances within and between residues, as well as evolutionary information to present proteins as residue-level graphical representations in DeepGlycanSite. Combined with Transformer blocks with self-attention mechanism to enhance feature extraction and complex relationship discovery.
The team leveraged geometric features, such as directions and distances within and between residues, as well as evolutionary information to present proteins as residue-level graphical representations in DeepGlycanSite. Combined with Transformer blocks with self-attention mechanism to enhance feature extraction and complex relationship discovery.
Researchers compared DeepGlycanSite to current state-of-the-art computational methods on an independent test set involving more than a hundred unique sugar-binding proteins. The results show that the average Matthews correlation coefficient (MCC) of DeepGlycanSite (0.625) is more than 30 times that of StackCBPred (0.018), and far exceeds other sequence-based prediction methods.
Traditional ligand binding site methods may exclude binding sites for simple sugar molecules due to hydrophobicity or small size, while DeepGlycanSite can effectively identify these sites.在 icon: Compare models to predict the performance of different sugar binding sites. (Source: Paper)
Moreover, DeepGlycanSite also performs well in predicting multiple sugar-binding sites on proteins, which is of great value for understanding how multivalent glycoconjugates affect sugar-protein interactions and the regulation of biological processes. For example, multivalent glycoconjugates are designed as chemical tools and drug candidates to influence the interaction between sugar molecules and lectins.
Different from traditional methods that only use protein sequence or structural information, DeepGlycanSite fully considers the geometric information and evolutionary characteristics of the protein, which may be a key factor in its excellent performance.
Additionally, given the chemical structure of a query sugar molecule, DeepGlycanSite can predict its specific binding site.的 icon: Query of the specific binding site prediction of sugar. (Source: Paper)
 Researchers explored the application of DeepGlycanSite to functionally important G protein-coupled receptors (GPCRs). Using the protein structure and carbohydrate chemical structure predicted by AlphaFold2, DeepGlycanSite successfully detected the specific binding site of GDP-Fuc on human P2Y14. Illustration: Experimental verification of DeepGlycanSite. (Source: Paper)
Researchers explored the application of DeepGlycanSite to functionally important G protein-coupled receptors (GPCRs). Using the protein structure and carbohydrate chemical structure predicted by AlphaFold2, DeepGlycanSite successfully detected the specific binding site of GDP-Fuc on human P2Y14. Illustration: Experimental verification of DeepGlycanSite. (Source: Paper)
While the quality of AlphaFold2’s predicted side chains could be improved, DeepGlycanSite relies less on protein structure accuracy and is able to use predicted protein structures to provide insights into sugar-protein interactions.
In summary, the validation of DeepGlycanSite in independent test sets and in vitro case studies shows that it is an effective tool for sugar binding site prediction. Researchers can use DeepGlycanSite to predict sugar-binding pockets on target proteins, thereby advancing understanding of sugar-protein interactions. Saccharides play a key role in biological functions. DeepGlycanSite not only helps analyze the biological functions of sugar molecules and sugar-binding proteins, but also provides a powerful tool for the development of sugar drugs.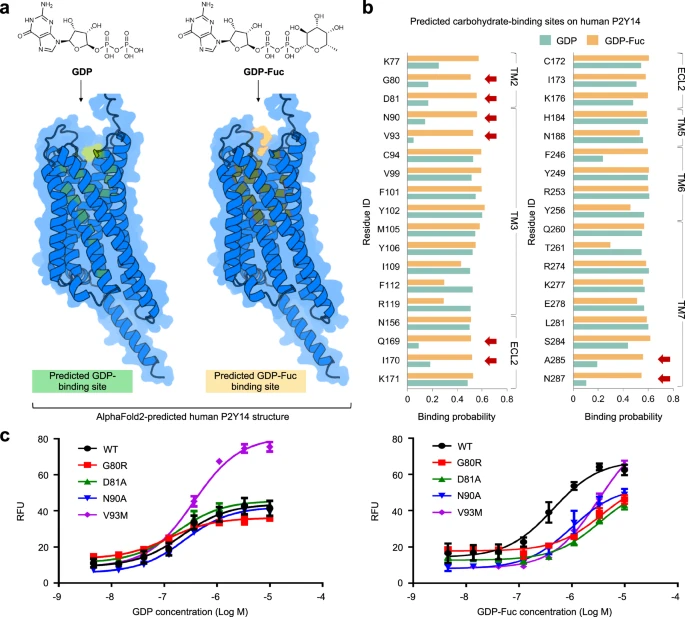
The above is the detailed content of 30 times more efficient than traditional methods, the Transformer deep learning model of the Chinese Academy of Sciences team predicts sugar-protein interaction sites. For more information, please follow other related articles on the PHP Chinese website!
 How to solve the problem of missing ssleay32.dll
How to solve the problem of missing ssleay32.dll
 How to open the terminal window in vscode
How to open the terminal window in vscode
 Is A5 bigger or B5 paper bigger?
Is A5 bigger or B5 paper bigger?
 The role of parseint function
The role of parseint function
 The difference between arrow functions and ordinary functions
The difference between arrow functions and ordinary functions
 How to define an array
How to define an array
 How much is Snapdragon 8gen2 equivalent to Apple?
How much is Snapdragon 8gen2 equivalent to Apple?
 fil currency price real-time price
fil currency price real-time price
 SpringBoot project building steps
SpringBoot project building steps








![[Web front-end] Node.js quick start](https://img.php.cn/upload/course/000/000/067/662b5d34ba7c0227.png)



