


Deep learning methods have promoted significant progress in single-state prediction of biomolecular structures. However, the functionality of biomolecules depends on the range of conformations they can assume. This is especially true for peptides, a class of highly flexible molecules that participate in a variety of biological processes and are of great interest as therapeutics.
Philip M. Kim and Osama Abdin at the University of Toronto developed PepFlow, a transferable generative model that enables all-atom sampling directly from the allowed conformational space of an input peptide. The researchers trained the model in a diffusion framework and then used equivalent flow for conformational sampling.
To overcome the prohibitive cost of generalized all-atom modeling, they modularized the generation process and integrated supernetworks to predict sequence-specific network parameters. PepFlow accurately predicts peptide structures and efficiently reproduces experimental peptide collections in a fraction of the run time of traditional methods. PepFlow can also be used to sample conformations that satisfy constraints such as macrocyclization.
"So far, we have not been able to simulate the full conformation of a peptide." Osama Abdin, the first author of the study, said, "PepFlow uses deep learning to capture the precise conformation of the peptide in minutes. This model has the potential to be used by design Peptides as binders to guide drug development"
The study was titled "Direct conformational sampling from peptide energy landscapes through hypernetwork-conditioned diffusion" and was published in "Nature Machine Intelligence" on June 27, 2024.

Therapeutic Potential of Peptides
Peptide Modeling and Engineering
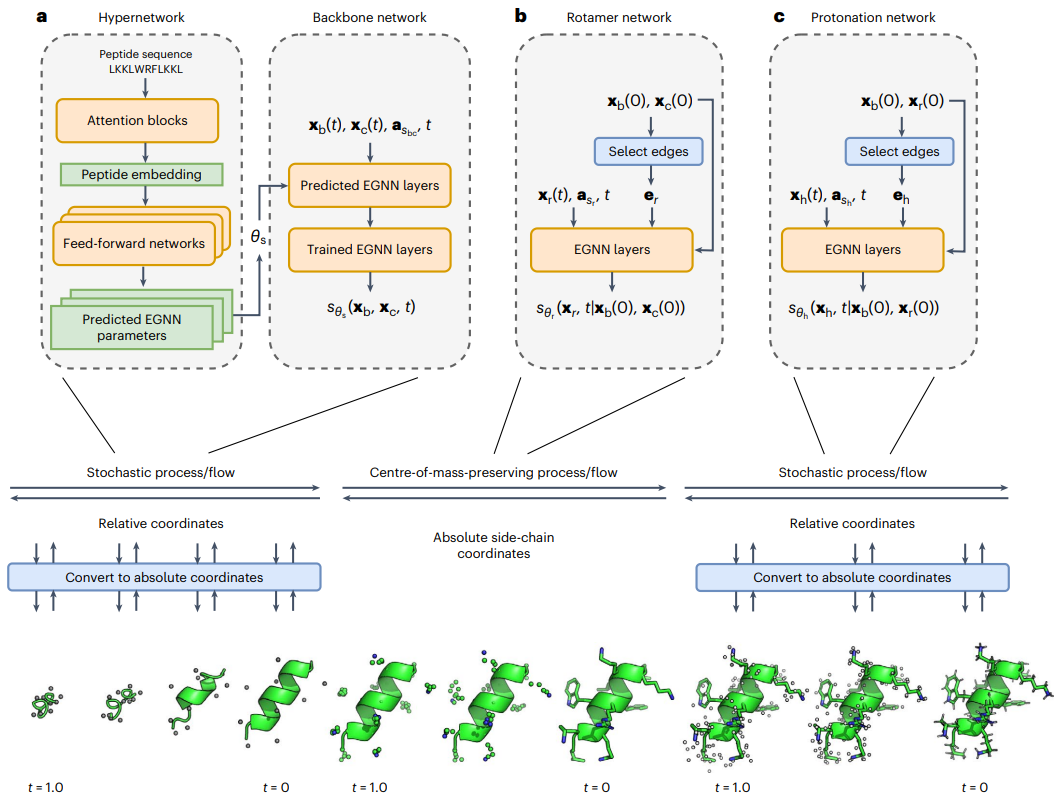
To solve this problem, they developed PepFlow, a modular, hypernetwork conditional generative model that can predict all-atom conformations for any input peptide sequence. PepFlow is a continuous-time diffusion model trained on known molecular conformations. The corresponding probabilistic flow ODE is used for energy sampling and training.
PepFlow has powerful capabilities for predicting singlet peptide structures and collections of short linear motifs (SLiM), and can model peptide structures under constraints such as macrocyclization through latent space conformation searches.
This model extends the ability of AlphaFold, the leading Google Deepmind AI system, to predict protein structures. PepFlow outperforms AlphaFold2 by generating a range of conformations for a given peptide; AlphaFold2 was not designed to solve this problem.
What sets PepFlow apart is the technological innovation behind it. For example, it is a generalized model inspired by the Boltzmann generator, a very advanced physics-based machine learning model.
“Using PepFlow modeling can provide insight into the true energy status of peptides.” Abdin said, “It took two and a half years to develop PepFlow and only one month to train it, but it is worth moving to the next frontier and beyond. Models that predict only one structure of a peptide. Overall, the ability to accurately and efficiently sample peptide conformations has the potential to improve peptide docking and design. Peptide docking methods typically start with a library of peptide conformations docked to the protein of interest. More precise generation of peptide ensembles may improve this process.
PepFlow can also be used to evaluate the propensity of different sequences to assume conformations at target protein-protein interfaces, which can in turn be used to design inhibitory peptides.
Illustration: Comparison of ensembles generated by PepFlow and molecular dynamics simulations. (Source: paper) 
PepFlow has a significant drawback, unlike the Boltzmann generator, PepFlow lacks the ability to reweight the generated samples to achieve an accurate Boltzmann distribution.
While PepFlow is capable of performing likelihood calculations on generated samples, tractable calculations require the use of stochastic estimators, which adds noise to the calculated values. Additionally, PepFlow occasionally generates high-energy samples but is unable to capture the full energy landscape observed in molecular dynamics simulations.
One potential way to improve PepFlow is to transfer the developed model to other sampling frameworks. A normalized flow was used in the conditional settings and different sampling methods were used to facilitate sampling from the Boltzmann distribution.
The flow matching paradigm recently developed by the academic community further serves as an alternative to training continuous normalized flow models in a simulation-free manner. Flow matching has been effectively used for structural sampling of different molecules, including small molecules and proteins, and can potentially be used to extend the effectiveness of the PepFlow framework.
In summary, PepFlow is designed to be easily extensible to account for other factors, new information, and potential uses.
Even as a first version, PepFlow is a comprehensive and effective model with potential for further development of therapeutics that rely on peptide binding to activate or inhibit biological processes.
Paper link: https://www.nature.com/articles/s42256-024-00860-4
Related reports: https://phys.org/news/2024-06-deep-outperforms-google-ai -peptide.html
The above is the detailed content of Nature sub-journal, better than AlphaFold, all-atom sampling, an AI method for predicting peptide structure. For more information, please follow other related articles on the PHP Chinese website!
















![[Web front-end] Node.js quick start](https://img.php.cn/upload/course/000/000/067/662b5d34ba7c0227.png)



