

Applications and challenges of molecular descriptors
Molecular descriptors are widely used in molecular modeling. However, in the field of AI-assisted molecular discovery, there is a lack of naturally applicable, complete, and original molecular representations, affecting model performance and interpretability.
Proposal of t-SMILES framework
The fragment-based multi-scale molecular characterization framework t-SMILES solves the problem of molecular characterization. The framework uses SMILES type strings to describe molecules and supports sequence models as generative models.
t-SMILES’ code algorithms
t-SMILES has three code algorithms: TSSA, TSDY and TSID.
Experimental results
Experiments show that the molecules generated by the t-SMILES model have 100% theoretical validity and high novelty, which is better than the model based on SOTA SMILES.
Furthermore, the t-SMILES model avoids overfitting and maintains similarity on labeled low-resource datasets while achieving higher novelty.
Published information
The study, titled "t-SMILES: a fragment-based molecular representation framework for de novo ligand design", was published in "Nature Communications" on June 11.

Research on molecular representation method based on SMILES
Effective characterization of molecules is a key factor affecting the performance of artificial intelligence models.
Graph Neural Networks (GNN) are popular for their ability to generate 100% efficient molecules, but their expressive capabilities are limited.
Simplified Molecular Linear Input Specification (SMILES), as a linear representation, is prone to producing chemically invalid strings. DeepSMILES and SELFIES are improvements as alternatives, but still have issues.
Furthermore, research shows that language models (LM) may outperform most GNNs in learning large, complex molecules. Recently, LMs based on Transformers have demonstrated their ability to generate text that closely resembles human writing.
Inspired by these ideas, the researchers chose SMILES as the starting choice for fragment description, and combined with advanced natural language processing technology to handle fragment-based molecular modeling tasks, which can fuse the graph model to pay more attention to molecular topology and LM The advantage of strong learning ability.
Generate 100% effective new molecules, better than SOTA
Therefore, the Hunan University team proposed a new molecular description framework based on fragmented molecules, t-SMILES (tree-based SMILES). The framework contains three t-SMILES encoding algorithms: TSSA (t-SMILES with shared atoms), TSDY (t-SMILES with virtual atoms but not IDs), and TSID (t-SMILES with IDs and virtual atoms).

The newly proposed t-SMILES framework
Compared with SMILES
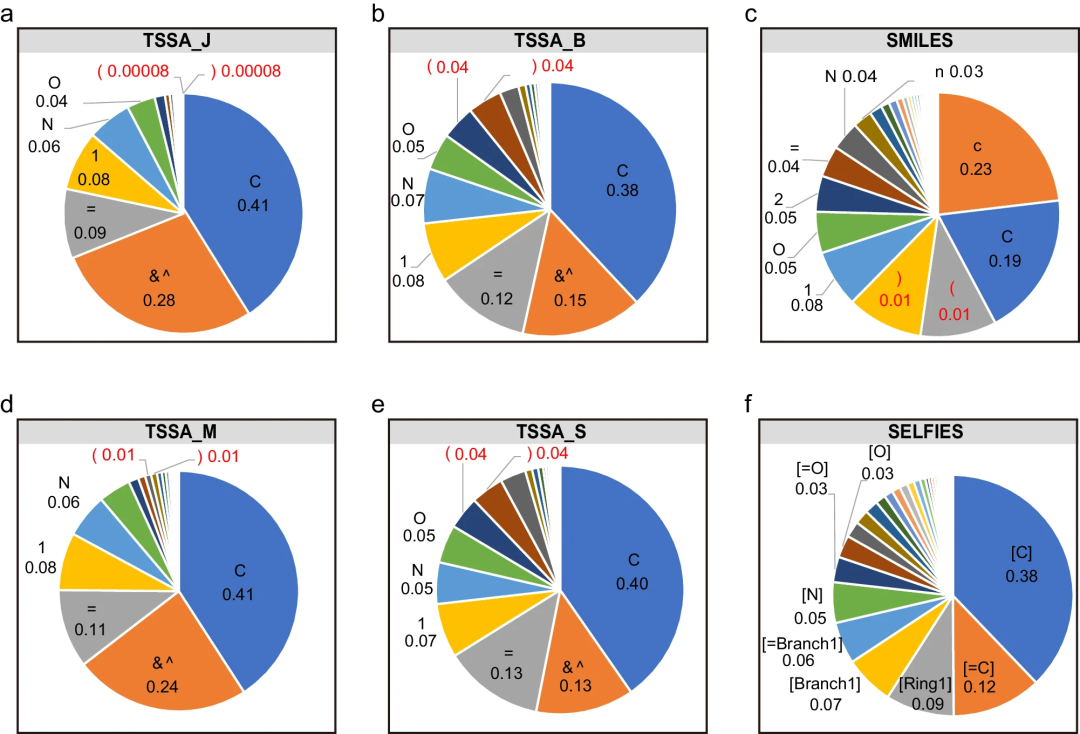
t-SMILES only introduces two new symbols "&" and "^" to encode multi-scale and hierarchical molecular topology.
t-SMILES algorithm
provides a scalable and adaptable framework that can theoretically support a wide range of substructure schemes.
The t-SMILES based model
is able to learn high-level topological structure information while processing detailed substructure information.
Multi-code system
t-SMILES algorithm can build a multi-code system for molecular description, where:
First, the researchers systematically evaluated t-SMILES by delving into its unique characteristics. Subsequently, experiments were conducted using TSSA and TSDY on two labeled low-resource datasets, JNK332 and AID170633.
The research focuses on the limitations of t-SMILES and its alternatives, which are achieved by leveraging standard, data augmentation and pre-trained fine-tuned models. Twenty goal-directed tasks on ChEMBL were evaluated in parallel using TSDY, TSSA, and TSID. Thorough experiments were also performed on ChEMBL, Zinc, and QM9 to compare t-SMILES and its alternatives by using similar setups. Furthermore, various fragment-based baseline models and SOTA GNN models are compared.
Finally, an ablation study is performed to confirm the effectiveness of the generative model based on SMILES with reconstruction. To evaluate the adaptability and flexibility of the t-SMILES algorithm, four previously published fragmentation algorithms were used to decompose molecules, including JTVAE, BRICS, MMPA, and Scaffold. Three metrics were used in different experiments: a distributed learning benchmark, a goal-directed benchmark, and the Wasserstein distance metric for physicochemical properties.
Detailed comparative experiments show that the new molecules generated by the t-SMILES model are 100% theoretically valid and better than the model based on SOTA SMILES. Compared to SMILES, DSMILES, and SELFIES, the overall solution of t-SMILES can avoid overfitting problems and significantly improve balanced performance on low-resource datasets, whether using data augmentation or a pre-trained and then fine-tuned model.
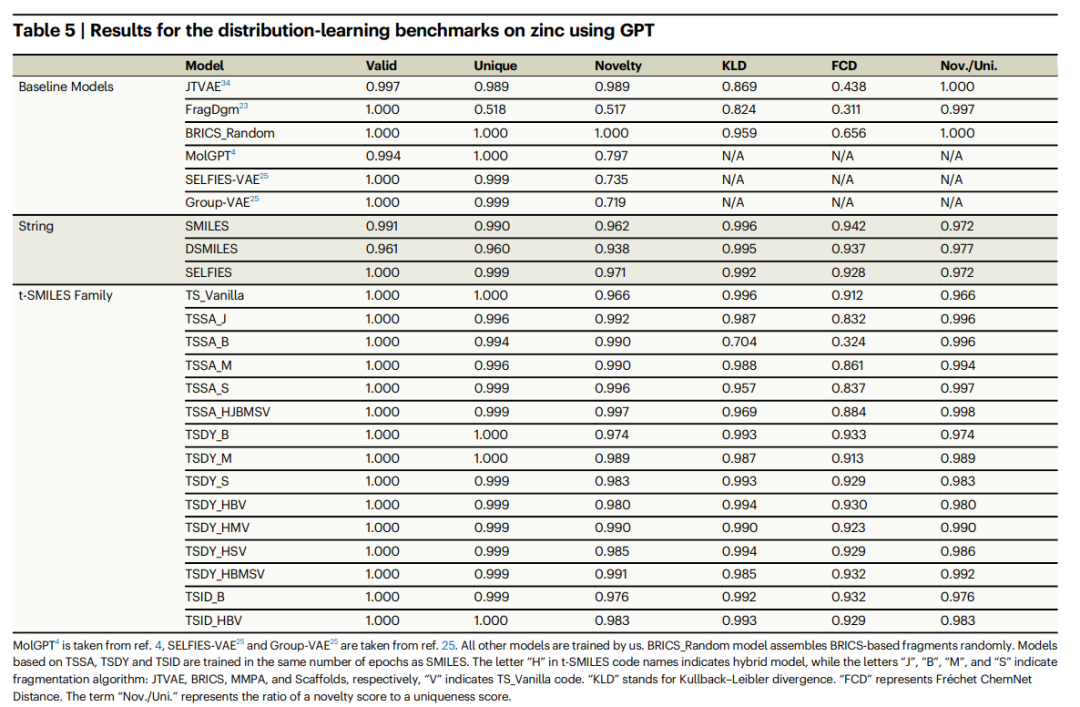
In addition, the t-SMILES model is able to skillfully capture the physicochemical properties of molecules, ensuring that the generated molecules maintain similarity with the training molecule distribution. This significantly improves performance compared to existing fragment-based and graph-based baseline models. In particular, the t-SMILES model with goal-oriented reconstruction algorithm shows clear advantages over SMILES, DSMILES, SELFIES, and SOTA CReM in goal-oriented tasks.
Limitations and room for improvement
Note: The cover comes from the Internet
The above is the detailed content of The molecule is 100% effective, ligands are designed from scratch, and Hunan University proposes a fragment-based molecular characterization framework. For more information, please follow other related articles on the PHP Chinese website!

















![[Web front-end] Node.js quick start](https://img.php.cn/upload/course/000/000/067/662b5d34ba7c0227.png)



