


단백질은 다른 분자와 결합하여 거의 모든 기본적인 생물학적 활동을 촉진합니다. 따라서 단백질 기능을 이해하는 것은 건강, 질병, 진화, 유기체 기능을 분자 수준에서 이해하는 데 중요합니다.
그러나 2억 개가 넘는 단백질은 특성이 밝혀지지 않은 상태로 남아 있으며, 계산 방법은 다양한 품질의 주석을 예측하기 위해 단백질의 구조 정보에 크게 의존합니다.
최근 옥스퍼드 대학교, ETH Zurich, 상하이 과학 기술 대학교, 베이징 사범 대학교의 연구팀은 기능 주석 및 단백질의 기능적 위치 식별을 촉진하기 위해 PhiGnet이라는 통계 기반 그래프 네트워크 방법을 설계했습니다.
PhiGnet은 성능 면에서 다른 방법보다 뛰어날 뿐만 아니라 구조적 정보가 없는 경우에도 시퀀스-기능 격차를 줄여줍니다. 연구 결과는 진화 데이터에 딥 러닝을 적용하면 잔류물 수준에서 기능적 위치를 강조하여 생물의학에서 단백질의 기존 특성과 새로운 기능을 해석하고 연구하는 데 귀중한 지원을 제공할 수 있음을 보여줍니다.
관련 연구는 "통계정보 그래프 네트워크를 활용한 단백질 기능의 정확한 예측"이라는 제목으로 "Nature Communications" 8월 4일자에 게재되었습니다.

단백질 기능을 이해하는 것은 많은 주요 생물학적 활동의 복잡한 메커니즘을 이해하는 데 중요하며 의학, 생명공학 및 의약품 개발 분야에는 광범위한 의미가 있습니다.
현재까지 UniProt 데이터베이스(2023년 6월)에서는 3억 5,600만 개가 넘는 단백질의 서열이 분석되었으며, 그 중 대다수(~80%)에는 알려진 기능 주석이 없습니다.
딥 러닝 방법은 ab initio 방법 및 상동성 모델링과 같은 고전적인 방법의 기능을 능가하여 단백질 3D 구조를 예측하는 데 놀라운 정확도를 달성했습니다. 그러나 단백질에 기능적 주석을 정확하게 할당하는 것은 특히 실험적 분석과 비교할 때 여전히 어려운 일입니다.
이러한 과제를 해결하기 위해 연구자들은 공동 진화하는 잔류물에 포함된 정보를 사용하여 잔류물 수준 기능에 주석을 달 수 있다는 가설을 세웠습니다.
옥스퍼드 대학교 팀은 통계 기반 그래프 네트워크를 사용하여 서열로만 단백질 기능을 예측할 것을 제안합니다. 이 방법은 본질적으로 진화적 특징을 특징으로 하며 특정 기능을 수행하는 잔류물의 중요성에 대한 정량적 평가를 허용합니다.
이 방법은 진화 데이터에서 얻은 지식을 활용하여 두 개의 누적 그래프 컨벌루션 네트워크를 구동합니다. 얻은 지식과 설계된 네트워크 아키텍처를 통해 단백질에 기능적 주석을 정확하게 할당할 수 있으며, 중요한 것은 특정 기능과 관련된 각 잔기의 중요성을 정량화할 수 있다는 것입니다.
단백질 기능 주석을 위한 PhiGnet
PhiGnet 방법은 통계 기반 그래프 네트워크를 사용하여 단백질 기능에 주석을 달고 서열을 기반으로 종 전체의 기능적 위치를 식별합니다.

진화 결합(EVC) 및 잔여 커뮤니티(RC)의 지식을 흡수하기 위해 연구원들은 누적 그래프 컨벌루션 네트워크(GCN)를 사용하는 이중 채널 아키텍처 접근 방식을 설계했습니다. 이 방법은 효소 위원회(EC) 번호 및 유전자 온톨로지(GO) 용어(생물학적 과정, BP, 세포 구성 요소, CC 및 분자 기능, MF)를 포함하여 단백질에 기능적 주석을 할당하도록 특별히 설계되었습니다.
단백질 서열이 제공되면 연구에서는 사전 훈련된 ESM-1b 모델을 사용하여 해당 서열의 임베딩을 도출합니다. 그 후, 임베딩은 EVC 및 RC(그래프 가장자리)뿐만 아니라 그래프 노드로서 듀얼 스택 GCN의 6개 그래프 컨벌루션 레이어에 입력됩니다. 이러한 레이어는 두 개의 완전히 연결된(FC) 레이어 블록과 함께 작동하여 두 GCN의 정보를 신중하게 처리하여 궁극적으로 단백질에 기능 주석을 할당하는 가능성을 평가하는 확률 텐서를 생성합니다.
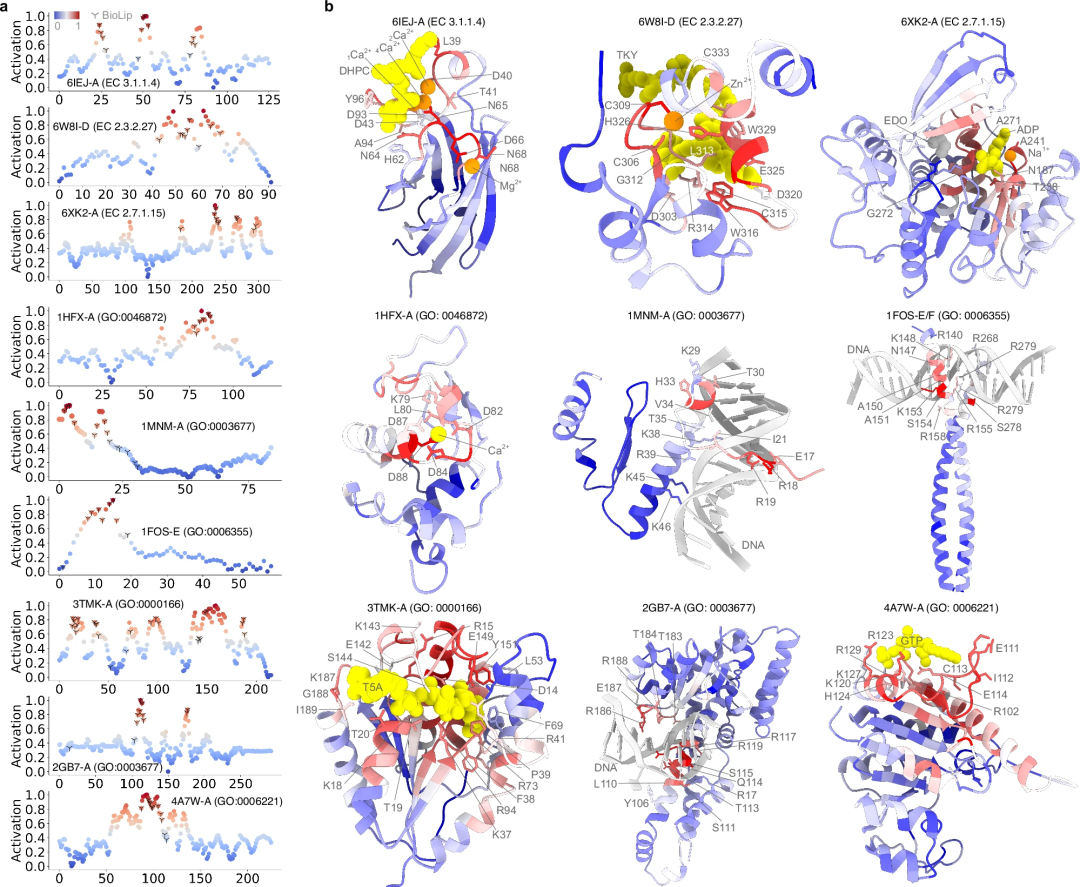
또한 Grad-CAM(Gradient-Weighted Class Activation Map) 방법을 사용하여 도출된 활성화 점수를 사용하여 특정 기능에서 각 잔기의 중요성을 평가합니다. 이 점수를 통해 PhiGnet은 개별 잔류물 수준에서 기능성 부위를 정확히 찾아낼 수 있습니다.
예를 들어, 세린-아스파르트산염 반복이 포함된 단백질 D(SdrD)의 RC를 계산하면 자연 진화를 통해 기능 부위의 잔기가 유지되는 것으로 나타났으며, PhiGnet은 이러한 정보를 포착할 수 있어 다음과 같은 분석이 향상됩니다. 잔기 구조적 데이터가 없는 경우에도 기본 수준에서 단백질 기능을 예측하는 방법입니다.
단백질 기능 부위에 주석을 답니다
Are computational predictions as accurate as experimentally determined functional annotations? To address this question, the study used activation scores to quantitatively examine the contribution of each amino acid to protein function. The predictive performance of PhiGnet was evaluated and the importance of residues (their contribution to protein function) in nine proteins was assessed.
Outperforms other state-of-the-art methods
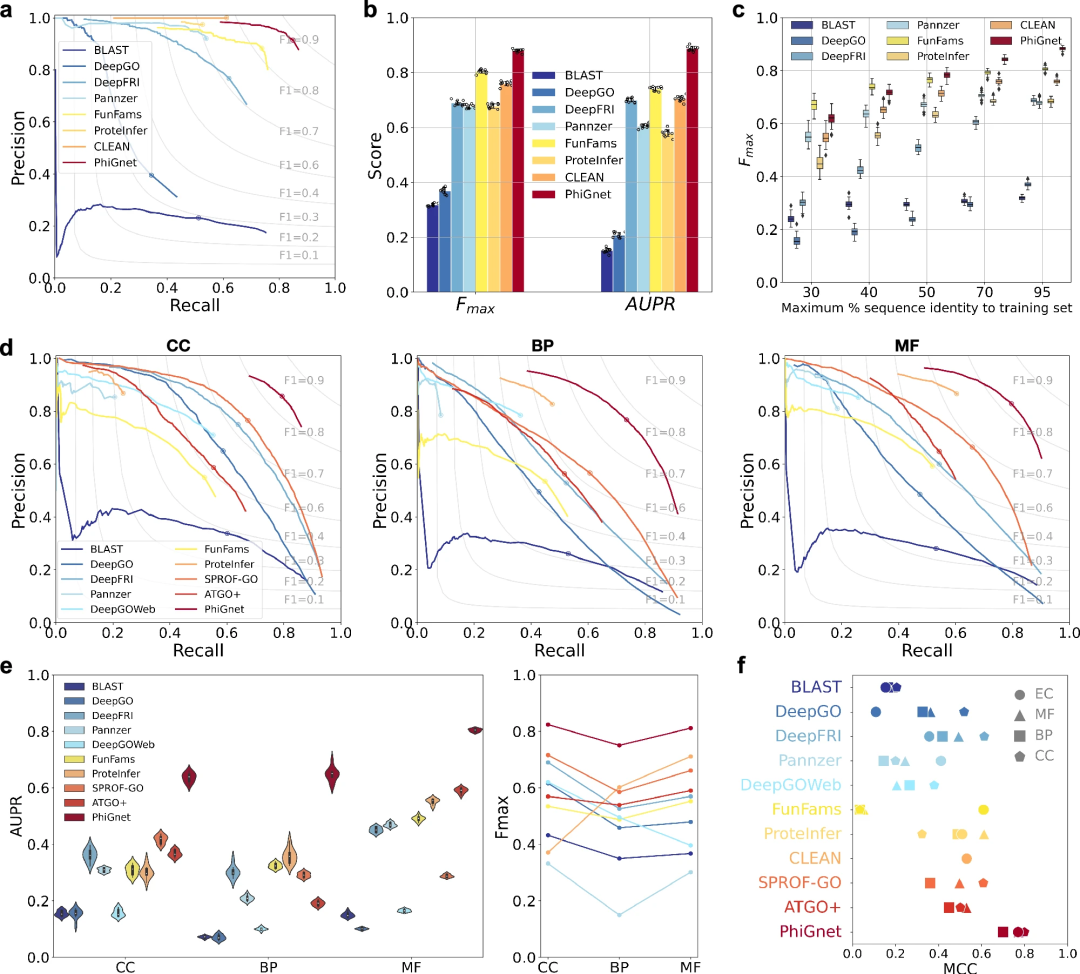
PhiGnet demonstrates the predictive power of assigning functional annotations to proteins in two test sets. It achieves average AUPR of 0.70 and 0.89, and Fmax scores of 0.80 and 0.88 for GO terms and EC numbers, respectively.
Overall, PhiGnet significantly outperforms all supervised and unsupervised methods on the benchmark dataset.
Additionally, the generalization robustness of PhiGnet was demonstrated to test proteins with different sequence identity thresholds than proteins in the training set. At different maximum sequence identity levels (30%, 40%, 50%, 70%, and 95%), PhiGnet showed better prediction performance as sequence identity increased.
Driven by evolutionary signatures
Evolutionary data plays an important role in PhiGnet and can be used to predict protein functional annotations and identify functional sites. First, ablation experiments were performed to test the contribution of EVC/RC to PhiGnet. Experiments show that PhiGnet can accurately assign protein functional annotations. Furthermore, PhiGnet using EVC or RC demonstrates a strong ability to learn general sequence-function relationships, often as well or as well as other methods.
Second, the ability of PhiGnet to characterize meaningful features from identified functionally relevant residues in residue communities was further investigated. Activation scores of residues were calculated to emphasize their contribution to protein function. Notably, the predicted residues are consistent with those of functional sites determined by experimental assays and are better identified than those in RC.
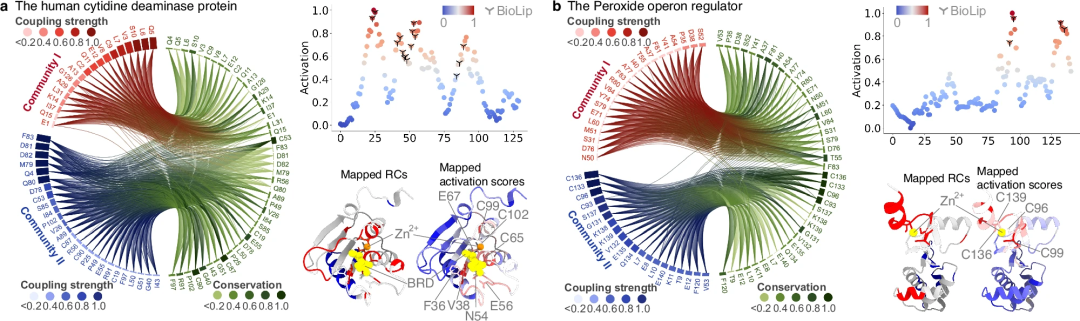
Research has shown that evolutionary information, especially the information contained in Remote Homology, is sufficient to specify the function of a protein and quantitatively characterize the residues of functional sites. Additionally, Remote Homology contains higher-order levels of evolutionary knowledge compared to the lower-order levels of information in the Evolutionary Vector. At the same time, the information contained in Remote Homology plays an important role in enhancing PhiGnet's ability to identify functionally relevant sites at the residue level.
Successes and Limitations
In summary, the better performance of PhiGnet can be attributed to its utilization of evolutionary data of protein sequences and higher-order patterns of the data, allowing for a deeper and more accurate understanding of protein function.
PhiGnet’s primary success is the use of statistical information graph convolutional neural networks to facilitate hierarchical learning of evolutionary data from massive sequence datasets. This approach significantly surpasses existing supervised and unsupervised methods and can be used to guide future biological and clinical experiments.
Limitations of the PhiGnet method include the bias/noise that occurs in protein families with low sequence diversity. Incorporating (co)evolutionary information into PhiGnet may affect the accurate identification of residue communities, especially if the information comes from highly conserved protein families. While integrating physically extracted knowledge into PhiGnet achieves significant improvements over other approaches, significant challenges remain in interpreting the learning mechanisms in PhiGnet.
The synergy between evolutionary data and machine learning will pave the way to accurately determine and engineer the biophysical properties of proteins.
The above is the detailed content of New SOTA for protein function prediction, statistics-based AI methods from Shanghai Institute of Technology, Oxford and others, published in Nature sub-journal. For more information, please follow other related articles on the PHP Chinese website!
 How to solve the problem of garbled characters when opening a web page
How to solve the problem of garbled characters when opening a web page
 What are the types of traffic?
What are the types of traffic?
 How to make a call without showing your number
How to make a call without showing your number
 What does dhcp mean?
What does dhcp mean?
 How to trigger keypress event
How to trigger keypress event
 gt540
gt540
 Can Douyin sparks be lit again if they have been off for more than three days?
Can Douyin sparks be lit again if they have been off for more than three days?
 How to connect to database using vb
How to connect to database using vb
 Server evaluation software
Server evaluation software








![[Web front-end] Node.js quick start](https://img.php.cn/upload/course/000/000/067/662b5d34ba7c0227.png)



