

According to foreign media Tech Xplore, researchers at MIT recently developed a new model called EquBind, which can predict the structure of new protein molecules in advance and improve the efficiency of drug development.
At present, this technology has been recognized by the industry, and papers describing this technology will also be accepted by the International Conference on Machine Learning (ICML) in July.
Currently, drug research and development is a long and expensive matter. The main reason is that developing drugs is very expensive. This cost includes not only billions of dollars in capital investment, but also decades of research time.
And during the research and development process, 90% of drugs will fail due to ineffectiveness or too many side effects. Only 10% of drugs can successfully pass the Food and Drug Administration inspection and be approved for marketing.
Therefore, pharmaceutical companies will increase the prices of successfully developed drugs to make up for the losses caused by failed drugs, so the prices of some drugs currently remain high.
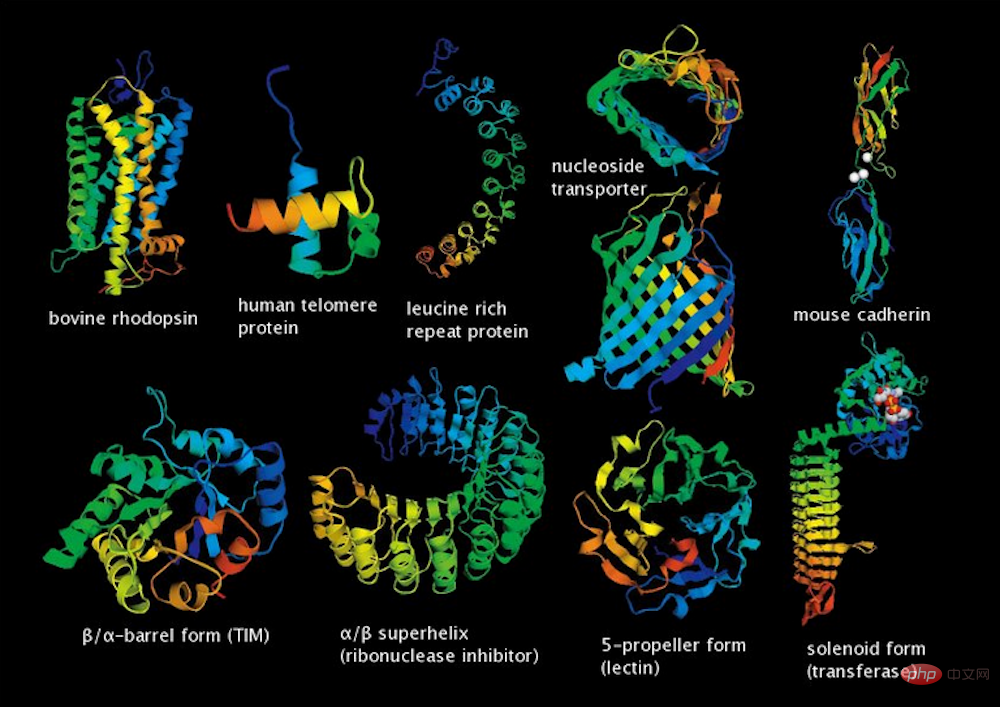
▲Some protein molecular structures
If researchers want to develop drugs, they must first find drug-like molecules with development potential. molecules). Another important reason for the slow progress of drug development is the huge number of existing drug-like molecules. Data show that there are currently as many as 1,016 existing drug-like molecules, a number that far exceeds the calculation upper limit of existing molecular calculation models.
In order to process such large molecules with data and speed up the process of drug development, Hannes St rk, a first-year graduate student in the Department of Electrical Engineering and Computer Science at MIT, developed a geometric deep learning model called "EquBind". EquBind runs 1,200 times faster than the fastest existing molecular computational docking models, allowing it to find drug-like molecules faster.
Currently, most traditional molecular computing docking models use a method called "ligand-protein" (ligand- to-protein binding) method to search for drug-like molecules. Specifically, the model needs to first receive a large number of sample molecules, then let the ligands bind to various molecules, and then the model scores different molecules, and then uses the final ranking to select the most suitable molecules. However, this approach has a complicated process and the model is less efficient in finding drug-like molecules.
Hannes St rk gave a vivid metaphor for this process. He said: "The previous typical 'ligand-protein' approach was like trying to get the model to insert the key into a lock with many keyholes. The model spends a lot of time scoring the fit between the key and each keyhole, and then selecting the most suitable one."
He continued to explain: "And EquBind can skip the most time-consuming steps. , can predict the most suitable 'keyhole' in advance when encountering new molecules, which is called 'blind docking'. EquBind has a built-in geometric reasoning algorithm that can help the model learn the basic structure of the molecule. This algorithm It allows EquBind to directly predict the most suitable position when encountering a new molecule, without spending a lot of time trying different positions and scoring."

▲Massachusetts Polytechnic
This model attracted the attention of Pat Walters, chief data officer of the therapeutic company Relay. Notice. Wolster suggested that Hannes Störk's research group use this model to develop drugs for lung cancer, leukemia and gastrointestinal tumors. Generally speaking, protein ligands used in drugs in these fields are difficult to dock using most traditional methods, but EquBind can successfully dock them.
▲Two inhibitor drugs to treat lung cancer
Walters said: "EquBind provides a unique solution to the protein docking problem. It Solving problems such as structure prediction and binding site identification. This method can make good use of thousands of public crystal structure information, and EquBind may impact the field in new ways."
Post this A paper on this technology will be accepted by the International Conference on Machine Learning (ICML) in July. Hannes Strk, the author of the paper, said: "I am looking forward to receiving some suggestions for improving the EquBind model at this conference."
AI pharmaceuticals is an emerging field that only entered the public eye in 2020.
The pharmaceutical field is a natural AI scenario. The long cycle, high cost, and low success rate of new drug research and development have left a huge place for AI to be used: machines can learn data independently, mine data, summarize drug research and development rules beyond expert experience, and then optimize the drug research and development process. In all aspects, this can not only improve the efficiency and success rate of drug research and development, but is also expected to reduce research and development expenses and trial and error costs.
Because of such characteristics and development potential, AI pharmaceuticals are currently gaining momentum. However, some people in the industry are pessimistic, saying that AI plays only a supporting role in the pharmaceutical process and cannot circumvent the inherent processes and mechanisms of the industry. It is impossible to complete ten years of work in two or three years.
But overall, there are still new technological breakthroughs in the field of AI pharmaceuticals, and development is booming.
The above is the detailed content of Efficiency increased by 1200 times! MIT develops new AI pharmaceutical model. For more information, please follow other related articles on the PHP Chinese website!

















![[Web front-end] Node.js quick start](https://img.php.cn/upload/course/000/000/067/662b5d34ba7c0227.png)



