
 Technology peripherals
AI
Stanford and Microsoft teamed up to use diffusion models to generate protein structures, which has been open source
Technology peripherals
AI
Stanford and Microsoft teamed up to use diffusion models to generate protein structures, which has been open source
Stanford and Microsoft teamed up to use diffusion models to generate protein structures, which has been open source

Proteins are vital to life and play a role in nearly every biological process. On the one hand, they can transmit signals between neurons, identify microscopic invaders, and activate immune responses. On the other hand, proteins have been extensively studied as a therapeutic mediator as part of the treatment of diseases. Thus, by generating new, physically foldable protein structures, the door is opened to new ways of exploiting cellular pathways to treat disease.
In this article, researchers from Stanford University, Microsoft Research and other institutions, inspired by the protein folding process in vivo, introduced a folding diffusion (folding diffusion, FoldingDiff) ) model, which designs protein backbone structures by mirroring the natural folding process of proteins.

- ##Paper address: https://arxiv .org/pdf/2209.15611.pdf
- Code address: https://github.com/microsoft/foldingdiff
Specifically, they describe the protein backbone structure as a series of consecutive angles that capture the relative orientation of the constituent amino acid residues, which The inherent displacement and rotation invariance of this representation greatly alleviates the need for complex equivariant networks.
This study trained a denoised diffusion probabilistic model based on the transformer backbone and demonstrated that our model can unconditionally generate highly realistic protein structures with complexity and structural patterns similar to those of natural protein.

Some netizens said: I wonder if this model will bring some competition to AlphaFold.

We can understand proteins as chains of amino acid residues of variable length. Typical amino acids are 20 species, sharing the same three-atom N-C_α-C backbone, but with different side chains connected to the C_α atoms (usually denoted as R, see Figure 1).
These residues assemble to form polymer chains that fold into a 3D structure, the shape of which largely determines the protein's function. These folded structures can be described using four levels:
- Primary structure, which simply captures the linear sequence of amino acids;
- Secondary structure describes the local arrangement of amino acids;
- tertiary structure describes the complete spatial arrangement of all residues;
- quaternary structure , describes how multiple different amino acid chains come together to form larger complexes.
The study proposes a simplified protein backbone framework that follows the biological process of protein folding while eliminating the need for complex equivariant networks. Rather than viewing a protein backbone, N amino acids in length, as a three-dimensional coordinate, they viewed it as a sequence of six internal, consecutive angles. That is, given the position of the current residue, the vector of six interior angles describes the relative positions of all backbone atoms in the next residue. These interior angles can be easily calculated using trigonometric functions, iteratively adding atoms to the protein backbone and then converting back to 3D Cartesian coordinates.

The picture below shows the results of an experiment. The Ramachandran diagram of the natural structure (figure a) contains three regions corresponding to the LH α-helix, RH α-helix, and β-sheet. All three regions are fully reproduced in the structure generated here (Fig. 3b). In other words, FoldingDiff is able to generate secondary structure elements within the protein backbone. Additionally, experiments show that the FoldingDiff model correctly learns that RH α-helices are more common than LH α-helices. Previous work using equivariant networks was unable to distinguish between these two types of spirals.

The following figure shows the secondary structure in the test main chain (4a) and the generated main chain (4b) Two-dimensional histogram, the results show that the generated structure reflects the true structure of the protein, with multiple α-helices, multiple β-sheets, and a mixture of the two.

The figure below shows that 111 of the 780 generated structures (14.2%) are designable, and their scTM scores ≥0.5 (Fig. 5a), which is higher than the value of 11.8% reported by Trippe et al. We also see that the generated main chains are more similar to the training examples and tend to have better designability (5b).

For more information, please read the original paper.
The above is the detailed content of Stanford and Microsoft teamed up to use diffusion models to generate protein structures, which has been open source. For more information, please follow other related articles on the PHP Chinese website!

Hot AI Tools

Undresser.AI Undress
AI-powered app for creating realistic nude photos

AI Clothes Remover
Online AI tool for removing clothes from photos.

Undress AI Tool
Undress images for free

Clothoff.io
AI clothes remover

AI Hentai Generator
Generate AI Hentai for free.

Hot Article
Hot Tools

Notepad++7.3.1
Easy-to-use and free code editor

SublimeText3 Chinese version
Chinese version, very easy to use

Zend Studio 13.0.1
Powerful PHP integrated development environment

Dreamweaver CS6
Visual web development tools

SublimeText3 Mac version
God-level code editing software (SublimeText3)
Hot Topics
 1378
1378
 52
52

 The world's most powerful open source MoE model is here, with Chinese capabilities comparable to GPT-4, and the price is only nearly one percent of GPT-4-Turbo
May 07, 2024 pm 04:13 PM
The world's most powerful open source MoE model is here, with Chinese capabilities comparable to GPT-4, and the price is only nearly one percent of GPT-4-Turbo
May 07, 2024 pm 04:13 PM
Imagine an artificial intelligence model that not only has the ability to surpass traditional computing, but also achieves more efficient performance at a lower cost. This is not science fiction, DeepSeek-V2[1], the world’s most powerful open source MoE model is here. DeepSeek-V2 is a powerful mixture of experts (MoE) language model with the characteristics of economical training and efficient inference. It consists of 236B parameters, 21B of which are used to activate each marker. Compared with DeepSeek67B, DeepSeek-V2 has stronger performance, while saving 42.5% of training costs, reducing KV cache by 93.3%, and increasing the maximum generation throughput to 5.76 times. DeepSeek is a company exploring general artificial intelligence
 AI subverts mathematical research! Fields Medal winner and Chinese-American mathematician led 11 top-ranked papers | Liked by Terence Tao
Apr 09, 2024 am 11:52 AM
AI subverts mathematical research! Fields Medal winner and Chinese-American mathematician led 11 top-ranked papers | Liked by Terence Tao
Apr 09, 2024 am 11:52 AM
AI is indeed changing mathematics. Recently, Tao Zhexuan, who has been paying close attention to this issue, forwarded the latest issue of "Bulletin of the American Mathematical Society" (Bulletin of the American Mathematical Society). Focusing on the topic "Will machines change mathematics?", many mathematicians expressed their opinions. The whole process was full of sparks, hardcore and exciting. The author has a strong lineup, including Fields Medal winner Akshay Venkatesh, Chinese mathematician Zheng Lejun, NYU computer scientist Ernest Davis and many other well-known scholars in the industry. The world of AI has changed dramatically. You know, many of these articles were submitted a year ago.
 Google is ecstatic: JAX performance surpasses Pytorch and TensorFlow! It may become the fastest choice for GPU inference training
Apr 01, 2024 pm 07:46 PM
Google is ecstatic: JAX performance surpasses Pytorch and TensorFlow! It may become the fastest choice for GPU inference training
Apr 01, 2024 pm 07:46 PM
The performance of JAX, promoted by Google, has surpassed that of Pytorch and TensorFlow in recent benchmark tests, ranking first in 7 indicators. And the test was not done on the TPU with the best JAX performance. Although among developers, Pytorch is still more popular than Tensorflow. But in the future, perhaps more large models will be trained and run based on the JAX platform. Models Recently, the Keras team benchmarked three backends (TensorFlow, JAX, PyTorch) with the native PyTorch implementation and Keras2 with TensorFlow. First, they select a set of mainstream
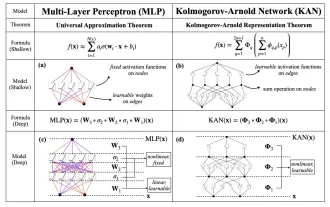 KAN, which replaces MLP, has been extended to convolution by open source projects
Jun 01, 2024 pm 10:03 PM
KAN, which replaces MLP, has been extended to convolution by open source projects
Jun 01, 2024 pm 10:03 PM
Earlier this month, researchers from MIT and other institutions proposed a very promising alternative to MLP - KAN. KAN outperforms MLP in terms of accuracy and interpretability. And it can outperform MLP running with a larger number of parameters with a very small number of parameters. For example, the authors stated that they used KAN to reproduce DeepMind's results with a smaller network and a higher degree of automation. Specifically, DeepMind's MLP has about 300,000 parameters, while KAN only has about 200 parameters. KAN has a strong mathematical foundation like MLP. MLP is based on the universal approximation theorem, while KAN is based on the Kolmogorov-Arnold representation theorem. As shown in the figure below, KAN has
 Hello, electric Atlas! Boston Dynamics robot comes back to life, 180-degree weird moves scare Musk
Apr 18, 2024 pm 07:58 PM
Hello, electric Atlas! Boston Dynamics robot comes back to life, 180-degree weird moves scare Musk
Apr 18, 2024 pm 07:58 PM
Boston Dynamics Atlas officially enters the era of electric robots! Yesterday, the hydraulic Atlas just "tearfully" withdrew from the stage of history. Today, Boston Dynamics announced that the electric Atlas is on the job. It seems that in the field of commercial humanoid robots, Boston Dynamics is determined to compete with Tesla. After the new video was released, it had already been viewed by more than one million people in just ten hours. The old people leave and new roles appear. This is a historical necessity. There is no doubt that this year is the explosive year of humanoid robots. Netizens commented: The advancement of robots has made this year's opening ceremony look like a human, and the degree of freedom is far greater than that of humans. But is this really not a horror movie? At the beginning of the video, Atlas is lying calmly on the ground, seemingly on his back. What follows is jaw-dropping
 Recommended: Excellent JS open source face detection and recognition project
Apr 03, 2024 am 11:55 AM
Recommended: Excellent JS open source face detection and recognition project
Apr 03, 2024 am 11:55 AM
Face detection and recognition technology is already a relatively mature and widely used technology. Currently, the most widely used Internet application language is JS. Implementing face detection and recognition on the Web front-end has advantages and disadvantages compared to back-end face recognition. Advantages include reducing network interaction and real-time recognition, which greatly shortens user waiting time and improves user experience; disadvantages include: being limited by model size, the accuracy is also limited. How to use js to implement face detection on the web? In order to implement face recognition on the Web, you need to be familiar with related programming languages and technologies, such as JavaScript, HTML, CSS, WebRTC, etc. At the same time, you also need to master relevant computer vision and artificial intelligence technologies. It is worth noting that due to the design of the Web side
 FisheyeDetNet: the first target detection algorithm based on fisheye camera
Apr 26, 2024 am 11:37 AM
FisheyeDetNet: the first target detection algorithm based on fisheye camera
Apr 26, 2024 am 11:37 AM
Target detection is a relatively mature problem in autonomous driving systems, among which pedestrian detection is one of the earliest algorithms to be deployed. Very comprehensive research has been carried out in most papers. However, distance perception using fisheye cameras for surround view is relatively less studied. Due to large radial distortion, standard bounding box representation is difficult to implement in fisheye cameras. To alleviate the above description, we explore extended bounding box, ellipse, and general polygon designs into polar/angular representations and define an instance segmentation mIOU metric to analyze these representations. The proposed model fisheyeDetNet with polygonal shape outperforms other models and simultaneously achieves 49.5% mAP on the Valeo fisheye camera dataset for autonomous driving
 Tesla robots work in factories, Musk: The degree of freedom of hands will reach 22 this year!
May 06, 2024 pm 04:13 PM
Tesla robots work in factories, Musk: The degree of freedom of hands will reach 22 this year!
May 06, 2024 pm 04:13 PM
The latest video of Tesla's robot Optimus is released, and it can already work in the factory. At normal speed, it sorts batteries (Tesla's 4680 batteries) like this: The official also released what it looks like at 20x speed - on a small "workstation", picking and picking and picking: This time it is released One of the highlights of the video is that Optimus completes this work in the factory, completely autonomously, without human intervention throughout the process. And from the perspective of Optimus, it can also pick up and place the crooked battery, focusing on automatic error correction: Regarding Optimus's hand, NVIDIA scientist Jim Fan gave a high evaluation: Optimus's hand is the world's five-fingered robot. One of the most dexterous. Its hands are not only tactile
