
 Technology peripherals
AI
AI alchemy revolutionizes chemistry: MIT scholars use generative AI to generate new chemical reactions in six seconds
Technology peripherals
AI
AI alchemy revolutionizes chemistry: MIT scholars use generative AI to generate new chemical reactions in six seconds
AI alchemy revolutionizes chemistry: MIT scholars use generative AI to generate new chemical reactions in six seconds


What needs to be rewritten is: Editor | Kaixia
Chemistry, starting from the ancient alchemy of "equivalent exchange", has always been a discipline that studies and controls the relationship between substances. Interacting disciplines. By continually unlocking and exploiting new chemical reactions, many new materials have been developed. These new materials not only bring convenience to people's lives, but also improve energy efficiency and promote sustainable development
A basic chemical reaction consists of reactants, transition states (TS), and products . Transition states are crucial 3D structures in chemistry and are widely used to understand chemical reaction mechanisms, estimate reaction energy barriers, and explore vast reaction networks. However, due to the extremely short time (femtosecond order) in which they exist during the reaction, it is almost impossible to isolate and characterize the transition state experimentally.
Rewritten content: Usually, people will use quantum chemical calculation methods to determine the transition state between known reactants and products by repeatedly solving the Schrödinger equation. However, this computational method is very expensive and notorious for its frequent failures. At the same time, this method is limited by personal experience, intuition and computational resources, and the chemical reactions that each person can explore are also limited. This limitation is particularly fatal when studying unknown complex reactions. It will cause researchers to ignore some potential reactions, thereby misjudging the reaction mechanism, thereby affecting the design of catalytic materials
In response to this problem, a group of researchers from the Massachusetts Institute of Technology (MIT) developed An alternative method based on machine learning was developed that can discover these structures in seconds. Their new model could assist chemists in exploring and designing new reactions and catalysts to generate useful products with high added value, such as fuel compounds or pharmaceuticals. In addition, the model is capable of simulating naturally occurring chemical reactions, such as those key to the evolution of life on early Earth.
Heather Kulik, professor of chemical engineering and chemistry at MIT, pointed out that understanding the specific structure of the transition state is very important for designing catalysts or understanding how natural systems perform certain transformations
Related research work is titled "Accurate "Transition state generation with an object-aware equivariant elementary reaction diffusion model" was published in the top international journal "Nature Computational Science".
Dr. Duan Chenru from MIT is the first author of the paper. Du Yuanqi, a doctoral student from Cornell University, Jia Haojun, a doctoral student from MIT, and Professor Heather Kulik from MIT are Co-author of the paper. Original link: [https://rdcu.be/dtGSF]

Please click the following link to view the paper: https://www.nature.com/articles/s43588- 023-00563-7
MIT News also reported on this research

Reporting link: https://news.mit. edu/2023/computational-model-captures-elusive-transition-states-1215
What needs to be rewritten is: Theoretical difficulties
Currently, chemists can Transition states are calculated using quantum chemical calculations based on density functional theory. However, this method requires a lot of computing resources, and it takes hours or even days to complete the calculation of a transition state
In order to solve the problem of long calculation time, some researchers have recently begun to try to use machine learning models to discover Transition state structure. However, almost all models developed to date require that the two reactants be modeled as a whole, with the reactants maintaining a specific geometric configuration relative to each other. Any other possible configuration will be mistaken by the machine learning model as a new reaction
Dr. Duan Chenru said that if the reactant molecules are rotated, in principle, they can still experience the same before and after rotation chemical reaction. Just like when we talk about electrolyzing water, we only say that water is converted into oxygen and hydrogen under specific conditions, without describing the relative geometric positions of these molecules. But in traditional machine learning methods, the model will treat the reactions of reactants and products at different geometric positions as two different reactions. This makes machine learning training more difficult and accuracy will decrease
The diffusion model is a generative model that has been widely used in image processing. Recently, diffusion models have also been used to generate 3D molecular and protein structures, protein-ligand docking and structure-based drug design. In these applications, diffusion models use 3D special Euclidean group (SE(3)) graph neural networks (GNNs) to preserve the alignment, translational and rotational symmetries of molecules. However, elementary reactions consist of reactants, transition states, and products and follow the "object-aware" SE(3) symmetry. This is because the interaction between the three objects in the elementary reaction is not carried out in the 3D Euclidean space, but is a causal connection on the higher-dimensional electronic potential energy surface. Therefore, the existing diffusion model based on SE(3) GNN may have problems due to the destruction of symmetry
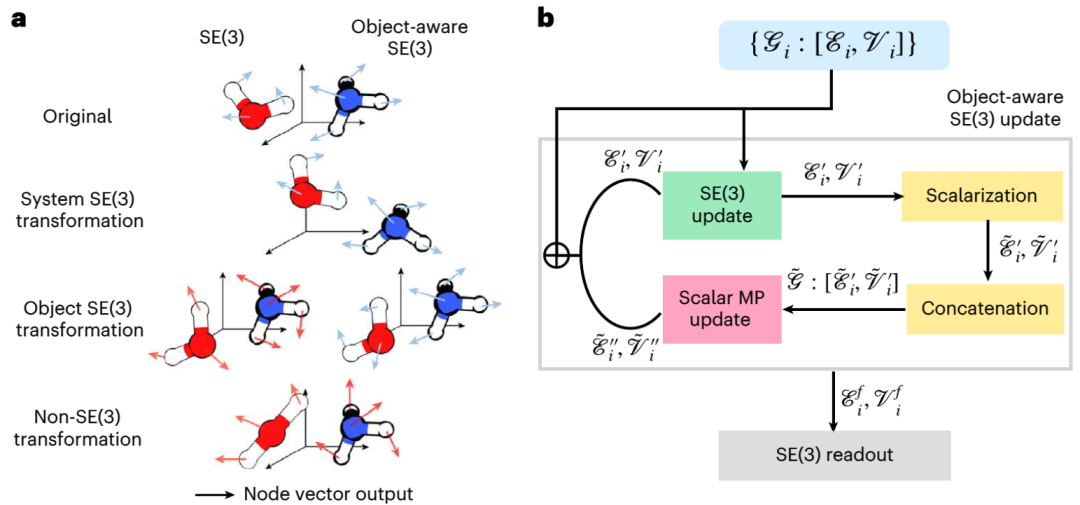
Illustration: "Object Perception" SE(3) etc. Variation and its implementation based on SE(3) equivariant GNN. (Source: paper)
Solution
The MIT team developed a new method called "OA-ReactDiff" based on the above problems. The team adapted the SE(3) equivariant GNN to "object-aware" simulation, that is, maintaining the SE(3) equivariance of individual objects while maintaining their independent interactions in Euclidean space
Dr. Duan Chenru said that the diffusion model is part of the field of generative artificial intelligence, which captures the transformation process between simple distributions and complex distributions through random processes. Once the model learns the basic distribution of how these three structures coexist, we can give it new reactants and products and it will try to generate transition state structures that correspond to those reactants and products
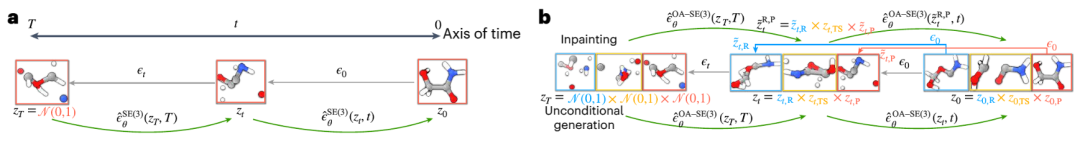
Illustration: Overview of the Equivariant Diffusion Model (EDM) used to generate samples of molecular systems. (Source: paper)
In the study, researchers used quantum computing methods to obtain the structures of reactants, transition states and products of 9,000 different chemical reactions in the training set. And also tested about 1,000 previously unseen reactions, requiring the generation of 40 possible structures for each transition state
In the calculation process, a "recommendation model" was introduced to predict which transition The highest confidence level. On this basis, further combined with uncertainty estimates, the researchers only performed quantum chemical calculations on the 14% of reactions with the highest model uncertainty, successfully achieving an average absolute error of 2.6 kcal/mol. This allows for results within an order of magnitude error when estimating reaction rates at 300°C using OA-ReactDiff. Compared with the transition state structure obtained by quantum chemical calculations, the root mean square error (RMSD) of the structure generated by OA-ReactDiff is in the range of 0.06 Angstroms (6 thousandths of a nanometer), an error magnitude that is almost indistinguishable to the naked eye
What is even more gratifying is that OA-ReactDiff only takes 6 seconds to generate a transition state structure, which is at least 1000 times faster than quantum chemical calculations. As a result, the algorithm successfully achieves extremely high accuracy and rapidity in calculating TS structures and reaction energy barriers.
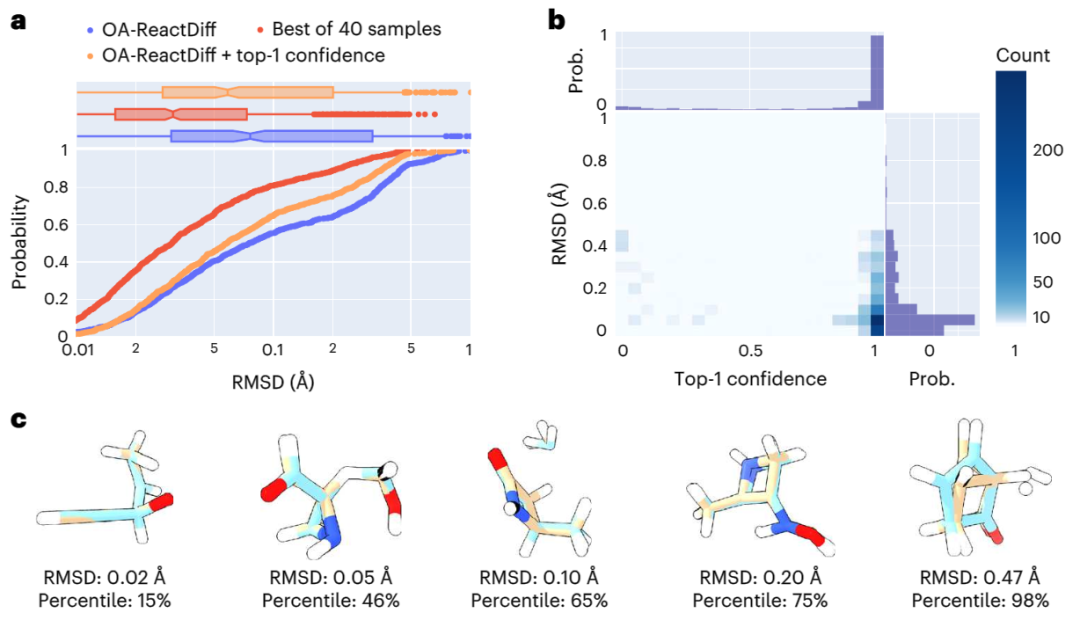
Illustration: Evaluating the structural similarity between the TS structure generated by OA-ReactDiff and the real TS structure. (Source: paper)
Professor Kulik also lamented, "It was difficult for us to imagine that thousands of transition states can be generated in one thought."
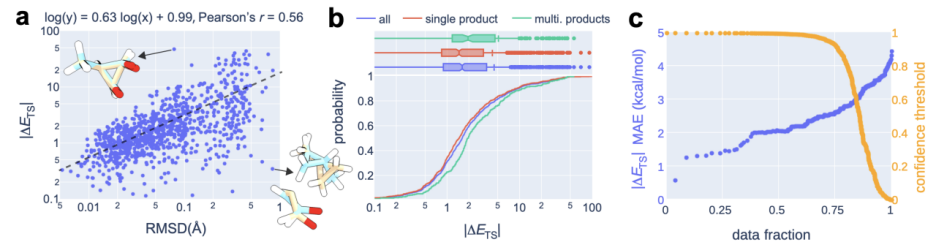
The content that needs to be rewritten is: Illustration: OA-ReactDiff plus recommends the energy performance of the TS structure. (Source: Paper)
Future expectations are expectations and hopes for the future. It is a person's vision for his or her future development and growth. Everyone has their own future expectations, which can be to achieve personal goals, pursue career success, build a happy family, or make positive contributions to society, etc. No matter what the future expectations are, it is the motivation and direction for people to work hard. Through hard work and persistence, we can gradually realize our future expectations and make our lives better and more fulfilling
What needs to be rewritten is: This research is the first to use 3D in chemical reactions Diffusion model. The significance of this work cannot be ignored, although the researchers only studied compounds with smaller numbers of atoms (
Professor Kulik pointed out: "Even when faced with larger systems or even enzyme-catalyzed systems, it is still possible to obtain different ways in which the atoms are most likely to rearrange."
The researchers now plan to extend their model by adding other components, such as catalysts. Leveraging the randomness of generative AI, OA-ReactDiff can explore unexpected chemical reactions. This feature complements the existing chemistry-based intuitive reaction exploration framework, helps establish a more complete chemical reaction network, and assists in the research and development of new catalytic materials. Research in this area can help them accelerate the discovery of new catalysts for specific reactions. Additionally, their proposed algorithm could be useful for developing new processes for drugs, fuels, or other useful compounds, especially when the synthesis involves many chemical steps.
Dr. Duan Chenru pointed out that in the past, all these calculations were performed using quantum chemistry methods, but now we can replace quantum chemistry with faster generation models
The researchers also pointed out that chemical reactions It is the core of chemical research. In addition to catalyst design that is biased toward industrial applications, OA-ReactDiff also has many interesting potential applications, such as exploring gas interactions that may occur on other planets, simulating reaction processes during the evolution of early life on Earth, etc.
The above is the detailed content of AI alchemy revolutionizes chemistry: MIT scholars use generative AI to generate new chemical reactions in six seconds. For more information, please follow other related articles on the PHP Chinese website!

Hot AI Tools

Undresser.AI Undress
AI-powered app for creating realistic nude photos

AI Clothes Remover
Online AI tool for removing clothes from photos.

Undress AI Tool
Undress images for free

Clothoff.io
AI clothes remover

AI Hentai Generator
Generate AI Hentai for free.

Hot Article
Hot Tools

Notepad++7.3.1
Easy-to-use and free code editor

SublimeText3 Chinese version
Chinese version, very easy to use

Zend Studio 13.0.1
Powerful PHP integrated development environment

Dreamweaver CS6
Visual web development tools

SublimeText3 Mac version
God-level code editing software (SublimeText3)
Hot Topics



 Breaking through the boundaries of traditional defect detection, 'Defect Spectrum' achieves ultra-high-precision and rich semantic industrial defect detection for the first time.
Jul 26, 2024 pm 05:38 PM
Breaking through the boundaries of traditional defect detection, 'Defect Spectrum' achieves ultra-high-precision and rich semantic industrial defect detection for the first time.
Jul 26, 2024 pm 05:38 PM
In modern manufacturing, accurate defect detection is not only the key to ensuring product quality, but also the core of improving production efficiency. However, existing defect detection datasets often lack the accuracy and semantic richness required for practical applications, resulting in models unable to identify specific defect categories or locations. In order to solve this problem, a top research team composed of Hong Kong University of Science and Technology Guangzhou and Simou Technology innovatively developed the "DefectSpectrum" data set, which provides detailed and semantically rich large-scale annotation of industrial defects. As shown in Table 1, compared with other industrial data sets, the "DefectSpectrum" data set provides the most defect annotations (5438 defect samples) and the most detailed defect classification (125 defect categories
 NVIDIA dialogue model ChatQA has evolved to version 2.0, with the context length mentioned at 128K
Jul 26, 2024 am 08:40 AM
NVIDIA dialogue model ChatQA has evolved to version 2.0, with the context length mentioned at 128K
Jul 26, 2024 am 08:40 AM
The open LLM community is an era when a hundred flowers bloom and compete. You can see Llama-3-70B-Instruct, QWen2-72B-Instruct, Nemotron-4-340B-Instruct, Mixtral-8x22BInstruct-v0.1 and many other excellent performers. Model. However, compared with proprietary large models represented by GPT-4-Turbo, open models still have significant gaps in many fields. In addition to general models, some open models that specialize in key areas have been developed, such as DeepSeek-Coder-V2 for programming and mathematics, and InternVL for visual-language tasks.
 Google AI won the IMO Mathematical Olympiad silver medal, the mathematical reasoning model AlphaProof was launched, and reinforcement learning is so back
Jul 26, 2024 pm 02:40 PM
Google AI won the IMO Mathematical Olympiad silver medal, the mathematical reasoning model AlphaProof was launched, and reinforcement learning is so back
Jul 26, 2024 pm 02:40 PM
For AI, Mathematical Olympiad is no longer a problem. On Thursday, Google DeepMind's artificial intelligence completed a feat: using AI to solve the real question of this year's International Mathematical Olympiad IMO, and it was just one step away from winning the gold medal. The IMO competition that just ended last week had six questions involving algebra, combinatorics, geometry and number theory. The hybrid AI system proposed by Google got four questions right and scored 28 points, reaching the silver medal level. Earlier this month, UCLA tenured professor Terence Tao had just promoted the AI Mathematical Olympiad (AIMO Progress Award) with a million-dollar prize. Unexpectedly, the level of AI problem solving had improved to this level before July. Do the questions simultaneously on IMO. The most difficult thing to do correctly is IMO, which has the longest history, the largest scale, and the most negative
 Training with millions of crystal data to solve the crystallographic phase problem, the deep learning method PhAI is published in Science
Aug 08, 2024 pm 09:22 PM
Training with millions of crystal data to solve the crystallographic phase problem, the deep learning method PhAI is published in Science
Aug 08, 2024 pm 09:22 PM
Editor |KX To this day, the structural detail and precision determined by crystallography, from simple metals to large membrane proteins, are unmatched by any other method. However, the biggest challenge, the so-called phase problem, remains retrieving phase information from experimentally determined amplitudes. Researchers at the University of Copenhagen in Denmark have developed a deep learning method called PhAI to solve crystal phase problems. A deep learning neural network trained using millions of artificial crystal structures and their corresponding synthetic diffraction data can generate accurate electron density maps. The study shows that this deep learning-based ab initio structural solution method can solve the phase problem at a resolution of only 2 Angstroms, which is equivalent to only 10% to 20% of the data available at atomic resolution, while traditional ab initio Calculation
 Nature's point of view: The testing of artificial intelligence in medicine is in chaos. What should be done?
Aug 22, 2024 pm 04:37 PM
Nature's point of view: The testing of artificial intelligence in medicine is in chaos. What should be done?
Aug 22, 2024 pm 04:37 PM
Editor | ScienceAI Based on limited clinical data, hundreds of medical algorithms have been approved. Scientists are debating who should test the tools and how best to do so. Devin Singh witnessed a pediatric patient in the emergency room suffer cardiac arrest while waiting for treatment for a long time, which prompted him to explore the application of AI to shorten wait times. Using triage data from SickKids emergency rooms, Singh and colleagues built a series of AI models that provide potential diagnoses and recommend tests. One study showed that these models can speed up doctor visits by 22.3%, speeding up the processing of results by nearly 3 hours per patient requiring a medical test. However, the success of artificial intelligence algorithms in research only verifies this
 To provide a new scientific and complex question answering benchmark and evaluation system for large models, UNSW, Argonne, University of Chicago and other institutions jointly launched the SciQAG framework
Jul 25, 2024 am 06:42 AM
To provide a new scientific and complex question answering benchmark and evaluation system for large models, UNSW, Argonne, University of Chicago and other institutions jointly launched the SciQAG framework
Jul 25, 2024 am 06:42 AM
Editor |ScienceAI Question Answering (QA) data set plays a vital role in promoting natural language processing (NLP) research. High-quality QA data sets can not only be used to fine-tune models, but also effectively evaluate the capabilities of large language models (LLM), especially the ability to understand and reason about scientific knowledge. Although there are currently many scientific QA data sets covering medicine, chemistry, biology and other fields, these data sets still have some shortcomings. First, the data form is relatively simple, most of which are multiple-choice questions. They are easy to evaluate, but limit the model's answer selection range and cannot fully test the model's ability to answer scientific questions. In contrast, open-ended Q&A
 SOTA performance, Xiamen multi-modal protein-ligand affinity prediction AI method, combines molecular surface information for the first time
Jul 17, 2024 pm 06:37 PM
SOTA performance, Xiamen multi-modal protein-ligand affinity prediction AI method, combines molecular surface information for the first time
Jul 17, 2024 pm 06:37 PM
Editor | KX In the field of drug research and development, accurately and effectively predicting the binding affinity of proteins and ligands is crucial for drug screening and optimization. However, current studies do not take into account the important role of molecular surface information in protein-ligand interactions. Based on this, researchers from Xiamen University proposed a novel multi-modal feature extraction (MFE) framework, which for the first time combines information on protein surface, 3D structure and sequence, and uses a cross-attention mechanism to compare different modalities. feature alignment. Experimental results demonstrate that this method achieves state-of-the-art performance in predicting protein-ligand binding affinities. Furthermore, ablation studies demonstrate the effectiveness and necessity of protein surface information and multimodal feature alignment within this framework. Related research begins with "S
 Automatically identify the best molecules and reduce synthesis costs. MIT develops a molecular design decision-making algorithm framework
Jun 22, 2024 am 06:43 AM
Automatically identify the best molecules and reduce synthesis costs. MIT develops a molecular design decision-making algorithm framework
Jun 22, 2024 am 06:43 AM
Editor | Ziluo AI’s use in streamlining drug discovery is exploding. Screen billions of candidate molecules for those that may have properties needed to develop new drugs. There are so many variables to consider, from material prices to the risk of error, that weighing the costs of synthesizing the best candidate molecules is no easy task, even if scientists use AI. Here, MIT researchers developed SPARROW, a quantitative decision-making algorithm framework, to automatically identify the best molecular candidates, thereby minimizing synthesis costs while maximizing the likelihood that the candidates have the desired properties. The algorithm also determined the materials and experimental steps needed to synthesize these molecules. SPARROW takes into account the cost of synthesizing a batch of molecules at once, since multiple candidate molecules are often available
