
 Technology peripherals
AI
Predicting optimal options for synthesizing drug molecules using geometric deep learning methods, paving the way for new drug discovery
Technology peripherals
AI
Predicting optimal options for synthesizing drug molecules using geometric deep learning methods, paving the way for new drug discovery
Predicting optimal options for synthesizing drug molecules using geometric deep learning methods, paving the way for new drug discovery


Post-stage functionalization is an economical method for optimizing the properties of drug candidates. However, the chemical complexity of drug molecules often makes late-stage functionalization challenging
To solve this problem, researchers from the University of Munich, ETH Zurich, and Roche Basel collaborated to develop a late-stage Functionalization platform, which is based on geometric deep learning and high-throughput reaction screening technology
Given that borylation is one of the key steps of functionalization, we used computational models to predict the yield under different reaction conditions, The average absolute error range is 4-5%. The model was able to classify new reactions for known and unknown substrates with an accuracy of 92% and 67% respectively. We were able to accurately capture the regioselectivity of the main product, with an F-score of 67% for the classifier. We successfully identified many opportunities for structural diversification when applied to 23 different commercial drug molecules
The study is titled "Using Geometric Deep Learning to Enable High-Throughput Experiments to Promote Late-stage Drug Diversification", Published in the journal Nature Chemistry on November 23, 2023

LSF projects play an important role in medicinal chemistry research
The novelty and complexity of structures make synthesizing chemical target structures challenging when aiming to establish structure-activity relationships in medicinal chemistry. Structure-activity relationship models can guide lead compounds and lead compound optimization plans to improve the pharmacological activity and physicochemical properties of drug candidates. Efficient integration is crucial for the exploration of structure-activity relationships, which is the bottleneck of the design-make-test-analyze cycle
There are many alternative methods to activate and modify C-H bonds to achieve organic scaffolds Late Stage Functionalization (LSF), ranging from molecular building blocks to advanced drug molecules. Many catalytic systems provide directional and non-directional approaches, as well as chemical and site-selective access to modified analogues
Among the numerous LSF methods, the C-H borylation method is considered the most commonly used for fast compounds Diversify your approach. Organoboron compounds can be converted into a variety of functional groups as a reliable means for subsequent C-C bond coupling reactions, thereby enabling extensive structure-activity relationship research
However, currently, in drug discovery, LSF’s The app has only a few reports. Most of these reports focus on a single LSF reaction type. Direct LSF of multiple types of C–H bonds with different bond strengths, electronic properties, and steric and functional group environments poses challenges. Furthermore, conducting LSF projects is often time and resource intensive, which is not consistent with the tight schedules and limited assets of many medicinal chemistry projects
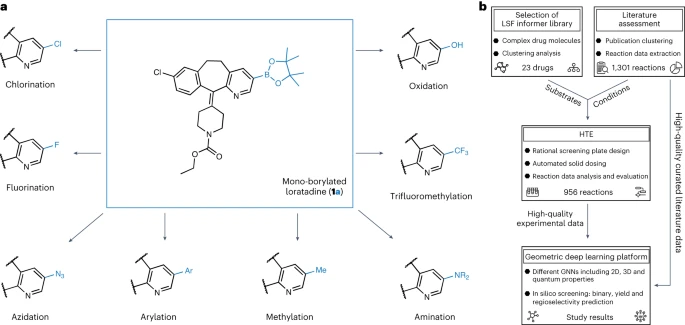
Diagram showing boronation An overview of diversity research. (Data source: paper)
Artificial Intelligence Supported LSF (Language Support Feature)
High-throughput experiment (HTE) is an established reaction optimization method. Semi-automated, miniaturized low-batch screening is possible, allowing multiple transformations to be performed in parallel using a small number of precious building blocks and consumables quickly and reproducibly. Combined with FAIR (Findability, Accessibility, Interoperability, Reusability) documentation that generates high-quality datasets on successful and failed responses, HTE enables advanced data analytics and machine learning to unlock LSF for drug discovery. Foundation.
Graph neural network (GNN) has been widely used in molecular feature extraction and attribute prediction. Among various machine learning methods developed for chemical reaction planning, GNNs have been successfully applied to retrosynthetic planning, regioselectivity prediction, and reaction product prediction. In addition, transformers and fingerprint-based methods have also been developed to solve similar problems
Research has shown that by learning the geometry of the transition state, the outcome of a competing reaction can be accurately predicted. Predictions of the regioselectivity of reactions driven by electronic effects can be improved using density functional theory (DFT) and graphical characterization of atomic partial charges. Combining graph machine learning and high-throughput experiments (HTE), conditions for C-H activation reactions of organic substrates can be optimized. Some studies have focused on the use of deep learning models of transition states that have the ability to predict reaction outcomes, including enantioselectivity in some cases. However, these methods are limited to small molecule structures and relatively small molecules. The small data set makes it challenging to apply such models to more structurally complex drug-like molecules. Based on literature research, the regioselectivity of iridium-catalyzed borylation reactions can be predicted through a hybrid machine learning model enhanced with quantum chemical information of transition states. However, the impact of steric and electronic effects has not yet been explored on the performance of C-H activation reaction models and their regioselective applications in molecules with multiple aromatic ring systems
Automated LSF Boron Screening with Geometric Deep Learning
Researchers from the University of Munich, ETH Zurich and Roche Pharmaceuticals Basel present an automatic LSF boron screening method applied to geometric deep learning LSF boron screening method for identifying late-stage hits and lead diversification opportunities. Computational deep learning was employed to predict reaction outcome, yield, and regioselectivity of complex drug molecules LSF.
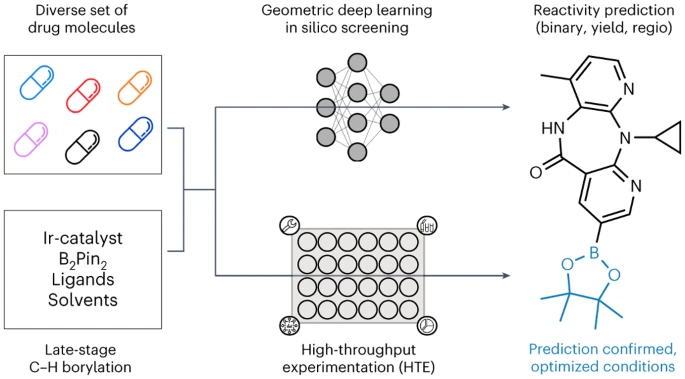
With this approach, the number of laboratory experiments could be significantly reduced, leading to a higher Efficiency and Sustainability of Chemical Synthesis
For the first step of this research, we conducted a thorough analysis of the published literature in order to select appropriate reaction conditions for high-throughput screening and late-stage lead compounds for drug discovery Substrates with relevant properties. We determined the reaction conditions based on a manually organized data set of 38 literatures
The selection of LSF substrates was based on the results of cluster analysis of 1,174 approved drugs, resulting in 23 structurally different drug molecules. This approach enables researchers to use relevant examples of reaction conditions and substrates in an "information library" approach, rather than relying solely on ideal substrates and fragments of limited applicability to optimize the synthesis of lead compounds
In the second step, researchers generate data (experimental datasets) using semi-automated high-throughput experiments (HTE). The reaction data of selected drug molecules and reaction conditions provide high-quality data for machine learning of subsequent reaction results
Finally, we trained different graph neural network (GNN) models using two-dimensional, three-dimensional and Molecular maps with enhanced partial charges on atoms to predict binary reaction outcomes, reaction yields, and regioselectivities. "Interestingly, the predictions improved when we considered three-dimensional information about the starting material rather than just its two-dimensional chemical formula," said Kenneth Atz, a PhD student at ETH Zurich. The method has been successfully used to identify positions in existing active ingredients where additional reactive groups can be introduced. This will help researchers more quickly develop new, more effective variants of known pharmaceutical active ingredients
Please click the following link to view the paper content: https://www.nature.com/articles/ s41557-023-01360-5
Related reports: https://techxplore.com/news/2023-11-Artificial intelligence paves the way for drugs.html
The above is the detailed content of Predicting optimal options for synthesizing drug molecules using geometric deep learning methods, paving the way for new drug discovery. For more information, please follow other related articles on the PHP Chinese website!

Hot AI Tools

Undresser.AI Undress
AI-powered app for creating realistic nude photos

AI Clothes Remover
Online AI tool for removing clothes from photos.

Undress AI Tool
Undress images for free

Clothoff.io
AI clothes remover

AI Hentai Generator
Generate AI Hentai for free.

Hot Article
Hot Tools

Notepad++7.3.1
Easy-to-use and free code editor

SublimeText3 Chinese version
Chinese version, very easy to use

Zend Studio 13.0.1
Powerful PHP integrated development environment

Dreamweaver CS6
Visual web development tools

SublimeText3 Mac version
God-level code editing software (SublimeText3)
Hot Topics
 1378
1378
 52
52

 DeepMind robot plays table tennis, and its forehand and backhand slip into the air, completely defeating human beginners
Aug 09, 2024 pm 04:01 PM
DeepMind robot plays table tennis, and its forehand and backhand slip into the air, completely defeating human beginners
Aug 09, 2024 pm 04:01 PM
But maybe he can’t defeat the old man in the park? The Paris Olympic Games are in full swing, and table tennis has attracted much attention. At the same time, robots have also made new breakthroughs in playing table tennis. Just now, DeepMind proposed the first learning robot agent that can reach the level of human amateur players in competitive table tennis. Paper address: https://arxiv.org/pdf/2408.03906 How good is the DeepMind robot at playing table tennis? Probably on par with human amateur players: both forehand and backhand: the opponent uses a variety of playing styles, and the robot can also withstand: receiving serves with different spins: However, the intensity of the game does not seem to be as intense as the old man in the park. For robots, table tennis
 The first mechanical claw! Yuanluobao appeared at the 2024 World Robot Conference and released the first chess robot that can enter the home
Aug 21, 2024 pm 07:33 PM
The first mechanical claw! Yuanluobao appeared at the 2024 World Robot Conference and released the first chess robot that can enter the home
Aug 21, 2024 pm 07:33 PM
On August 21, the 2024 World Robot Conference was grandly held in Beijing. SenseTime's home robot brand "Yuanluobot SenseRobot" has unveiled its entire family of products, and recently released the Yuanluobot AI chess-playing robot - Chess Professional Edition (hereinafter referred to as "Yuanluobot SenseRobot"), becoming the world's first A chess robot for the home. As the third chess-playing robot product of Yuanluobo, the new Guoxiang robot has undergone a large number of special technical upgrades and innovations in AI and engineering machinery. For the first time, it has realized the ability to pick up three-dimensional chess pieces through mechanical claws on a home robot, and perform human-machine Functions such as chess playing, everyone playing chess, notation review, etc.
 Claude has become lazy too! Netizen: Learn to give yourself a holiday
Sep 02, 2024 pm 01:56 PM
Claude has become lazy too! Netizen: Learn to give yourself a holiday
Sep 02, 2024 pm 01:56 PM
The start of school is about to begin, and it’s not just the students who are about to start the new semester who should take care of themselves, but also the large AI models. Some time ago, Reddit was filled with netizens complaining that Claude was getting lazy. "Its level has dropped a lot, it often pauses, and even the output becomes very short. In the first week of release, it could translate a full 4-page document at once, but now it can't even output half a page!" https:// www.reddit.com/r/ClaudeAI/comments/1by8rw8/something_just_feels_wrong_with_claude_in_the/ in a post titled "Totally disappointed with Claude", full of
 At the World Robot Conference, this domestic robot carrying 'the hope of future elderly care' was surrounded
Aug 22, 2024 pm 10:35 PM
At the World Robot Conference, this domestic robot carrying 'the hope of future elderly care' was surrounded
Aug 22, 2024 pm 10:35 PM
At the World Robot Conference being held in Beijing, the display of humanoid robots has become the absolute focus of the scene. At the Stardust Intelligent booth, the AI robot assistant S1 performed three major performances of dulcimer, martial arts, and calligraphy in one exhibition area, capable of both literary and martial arts. , attracted a large number of professional audiences and media. The elegant playing on the elastic strings allows the S1 to demonstrate fine operation and absolute control with speed, strength and precision. CCTV News conducted a special report on the imitation learning and intelligent control behind "Calligraphy". Company founder Lai Jie explained that behind the silky movements, the hardware side pursues the best force control and the most human-like body indicators (speed, load) etc.), but on the AI side, the real movement data of people is collected, allowing the robot to become stronger when it encounters a strong situation and learn to evolve quickly. And agile
 ACL 2024 Awards Announced: One of the Best Papers on Oracle Deciphering by HuaTech, GloVe Time Test Award
Aug 15, 2024 pm 04:37 PM
ACL 2024 Awards Announced: One of the Best Papers on Oracle Deciphering by HuaTech, GloVe Time Test Award
Aug 15, 2024 pm 04:37 PM
At this ACL conference, contributors have gained a lot. The six-day ACL2024 is being held in Bangkok, Thailand. ACL is the top international conference in the field of computational linguistics and natural language processing. It is organized by the International Association for Computational Linguistics and is held annually. ACL has always ranked first in academic influence in the field of NLP, and it is also a CCF-A recommended conference. This year's ACL conference is the 62nd and has received more than 400 cutting-edge works in the field of NLP. Yesterday afternoon, the conference announced the best paper and other awards. This time, there are 7 Best Paper Awards (two unpublished), 1 Best Theme Paper Award, and 35 Outstanding Paper Awards. The conference also awarded 3 Resource Paper Awards (ResourceAward) and Social Impact Award (
 Hongmeng Smart Travel S9 and full-scenario new product launch conference, a number of blockbuster new products were released together
Aug 08, 2024 am 07:02 AM
Hongmeng Smart Travel S9 and full-scenario new product launch conference, a number of blockbuster new products were released together
Aug 08, 2024 am 07:02 AM
This afternoon, Hongmeng Zhixing officially welcomed new brands and new cars. On August 6, Huawei held the Hongmeng Smart Xingxing S9 and Huawei full-scenario new product launch conference, bringing the panoramic smart flagship sedan Xiangjie S9, the new M7Pro and Huawei novaFlip, MatePad Pro 12.2 inches, the new MatePad Air, Huawei Bisheng With many new all-scenario smart products including the laser printer X1 series, FreeBuds6i, WATCHFIT3 and smart screen S5Pro, from smart travel, smart office to smart wear, Huawei continues to build a full-scenario smart ecosystem to bring consumers a smart experience of the Internet of Everything. Hongmeng Zhixing: In-depth empowerment to promote the upgrading of the smart car industry Huawei joins hands with Chinese automotive industry partners to provide
 Li Feifei's team proposed ReKep to give robots spatial intelligence and integrate GPT-4o
Sep 03, 2024 pm 05:18 PM
Li Feifei's team proposed ReKep to give robots spatial intelligence and integrate GPT-4o
Sep 03, 2024 pm 05:18 PM
Deep integration of vision and robot learning. When two robot hands work together smoothly to fold clothes, pour tea, and pack shoes, coupled with the 1X humanoid robot NEO that has been making headlines recently, you may have a feeling: we seem to be entering the age of robots. In fact, these silky movements are the product of advanced robotic technology + exquisite frame design + multi-modal large models. We know that useful robots often require complex and exquisite interactions with the environment, and the environment can be represented as constraints in the spatial and temporal domains. For example, if you want a robot to pour tea, the robot first needs to grasp the handle of the teapot and keep it upright without spilling the tea, then move it smoothly until the mouth of the pot is aligned with the mouth of the cup, and then tilt the teapot at a certain angle. . this
 Distributed Artificial Intelligence Conference DAI 2024 Call for Papers: Agent Day, Richard Sutton, the father of reinforcement learning, will attend! Yan Shuicheng, Sergey Levine and DeepMind scientists will give keynote speeches
Aug 22, 2024 pm 08:02 PM
Distributed Artificial Intelligence Conference DAI 2024 Call for Papers: Agent Day, Richard Sutton, the father of reinforcement learning, will attend! Yan Shuicheng, Sergey Levine and DeepMind scientists will give keynote speeches
Aug 22, 2024 pm 08:02 PM
Conference Introduction With the rapid development of science and technology, artificial intelligence has become an important force in promoting social progress. In this era, we are fortunate to witness and participate in the innovation and application of Distributed Artificial Intelligence (DAI). Distributed artificial intelligence is an important branch of the field of artificial intelligence, which has attracted more and more attention in recent years. Agents based on large language models (LLM) have suddenly emerged. By combining the powerful language understanding and generation capabilities of large models, they have shown great potential in natural language interaction, knowledge reasoning, task planning, etc. AIAgent is taking over the big language model and has become a hot topic in the current AI circle. Au
