

Editor| .
Although the controlled synthesis of substrate-catalyzed growth into carbon nanostructures is considered a promising approach, there are still challenges in dynamic catalytic surface growth mechanisms and design strategies that require further research and development.
Recently, a research team from Shanghai Jiao Tong University and Tohoku University in Japan demonstrated the effectiveness of an active machine learning model in revealing the microscopic process of substrate catalytic growth. Through the collaborative application of molecular dynamics and Monte Carlo methods, they successfully performed a comprehensive dynamic simulation of the growth of graphene on Cu(111). To enhance the accuracy of the simulation, the research team adopted a Gaussian approximation potential. This research provides new tools and methods for in-depth understanding of the catalytic growth process.
Through this research, we derived a practical and effective method that can be used to design metal or alloy substrates to obtain the desired carbon nanostructures and explore more reaction possibilities.
The research, titled "Active machine learning model for the dynamic simulation and growth mechanisms of carbon on metal surface", was published in "Nature Communications" on January 6, 2024.
 Paper link: https://www.nature.com/articles/s41467-023-44525-z
Paper link: https://www.nature.com/articles/s41467-023-44525-z
Substrate catalytic deposition is considered one of the most promising methods to achieve controllable growth of two- or three-dimensional covalently bonded networks of carbon atoms. While growth mechanisms on ordinary surfaces have been extensively studied, knowledge of the dynamic and atomic-scale factors that control graphene mass on high-index or composite surfaces is limited. This research gap has greatly hindered the development of theory-guided design approaches for novel catalytic metal substrates in the growth of carbon nanostructures.
Experimentally finding metal or alloy catalysts presents considerable challenges due to the wide range of potential substrates and the sensitivity of the carbon nanomaterial growth process to various experimental parameters.
Therefore, there is enough room for theoretical simulations and many atomic details are easily obtained. Examples include DFT, Kinetic Monte Carlo (KMC), and Ab initio Molecular Dynamics (AIMD). However, these methods each have their limitations. Therefore, there is still an urgent need for a robust design model that can accurately describe the carbon growth mechanism on metal surfaces.
Machine learning potentials (MLP) based on artificial neural networks or kernel methods are considered to be effective methods to solve the limited accuracy and transferability of classical force fields and maintain DFT-level accuracy. Despite significant achievements in data-driven MD simulations, constructing accurate MLPs remains a difficult task. One solution to this problem is dynamic learning techniques.
To improve the efficiency and effectiveness of dynamic training of deposition processes, a well-defined selection protocol is required. On the other hand, the dynamics of carbon growth on metallic substrates can be controlled by important rare events. Therefore, how to improve the training efficiency of MLP by combining boosted sampling methods with classical dynamics requires further research.
Data-driven automatic learning framework to generate MLP with minimum manpowerThis study proposes a data-driven automatic learning framework to generate MLP with minimum manpower , which is suitable for carbon growth on metal or alloy surfaces.
To achieve this task, the researchers used (1) Gaussian approximation potential (GAP) processing learning model; (2) an enhanced sampling method called timestamp force bias Monte Carlo (time-stamped force-biased Monte Carlo, tfMC) method to accelerate the relaxation process after carbon deposition, thereby including important rare events in the training database; (3) Smooth overlap based on atomic positions (SOAP) Efficient strategies for descriptor selection of representative training data; (4) well-established carbon training sets; (5) automated screening, fitting, and validation procedures.
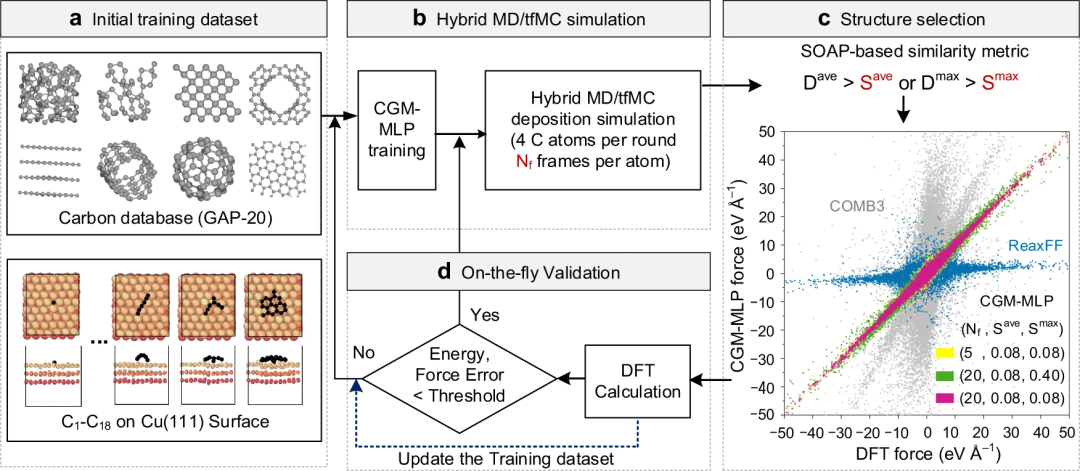 Figure 1: Carbon growth on metal generated by dynamic active learning machine learning potential (CGM) during a hybrid MD/tfMC simulation -MLP) schematic diagram. (Source: Paper)
Figure 1: Carbon growth on metal generated by dynamic active learning machine learning potential (CGM) during a hybrid MD/tfMC simulation -MLP) schematic diagram. (Source: Paper)
By exploiting the high accuracy of the Carbon Growth Machine Learning Potential (CGM-MLP) and incorporating rare atomic events in the MD/tfMC method, we successfully replicated graphene with metal surfaces The basic subprocesses related to nucleation and carbon growth are shown in the figure below.
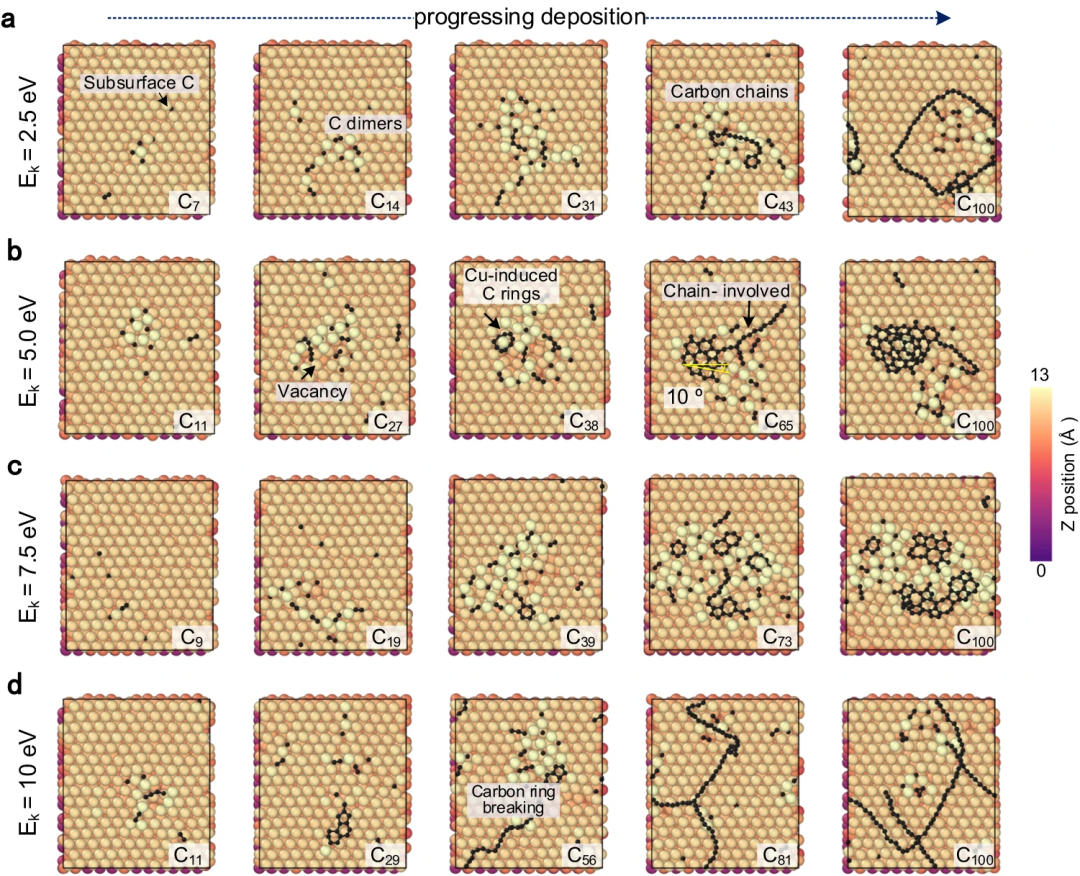
Figure 2: CGM-MLP driven simulation of graphene growth on Cu(111) with different carbon incident kinetic energies (Ek). (Source: Paper)
The resulting potential was then applied to study the deposition growth of carbon atoms on the Cu (111) surface. This method can correctly capture the key processes of carbon growth on Cu(111), such as the formation and migration of subsurface carbon monomers and surface dimers, the emergence of one-dimensional carbon nanocrystals, graphene nucleation involving Cu atoms and Edge passivation of carbon chains, and precipitation growth process.
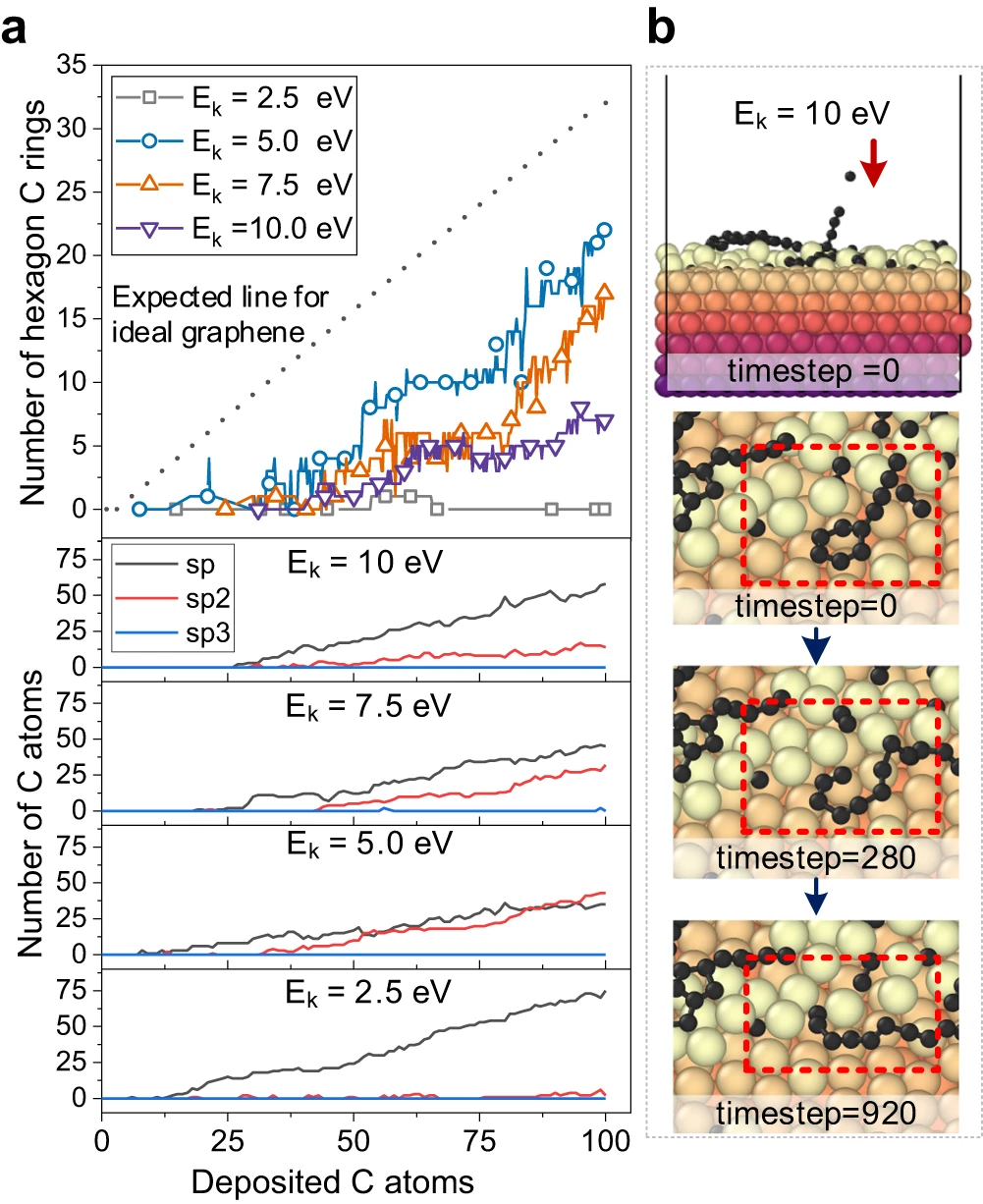
Figure 3: Carbon structure analysis and observation of carbon ring breakage by high-energy bombardment. (Source: Paper)
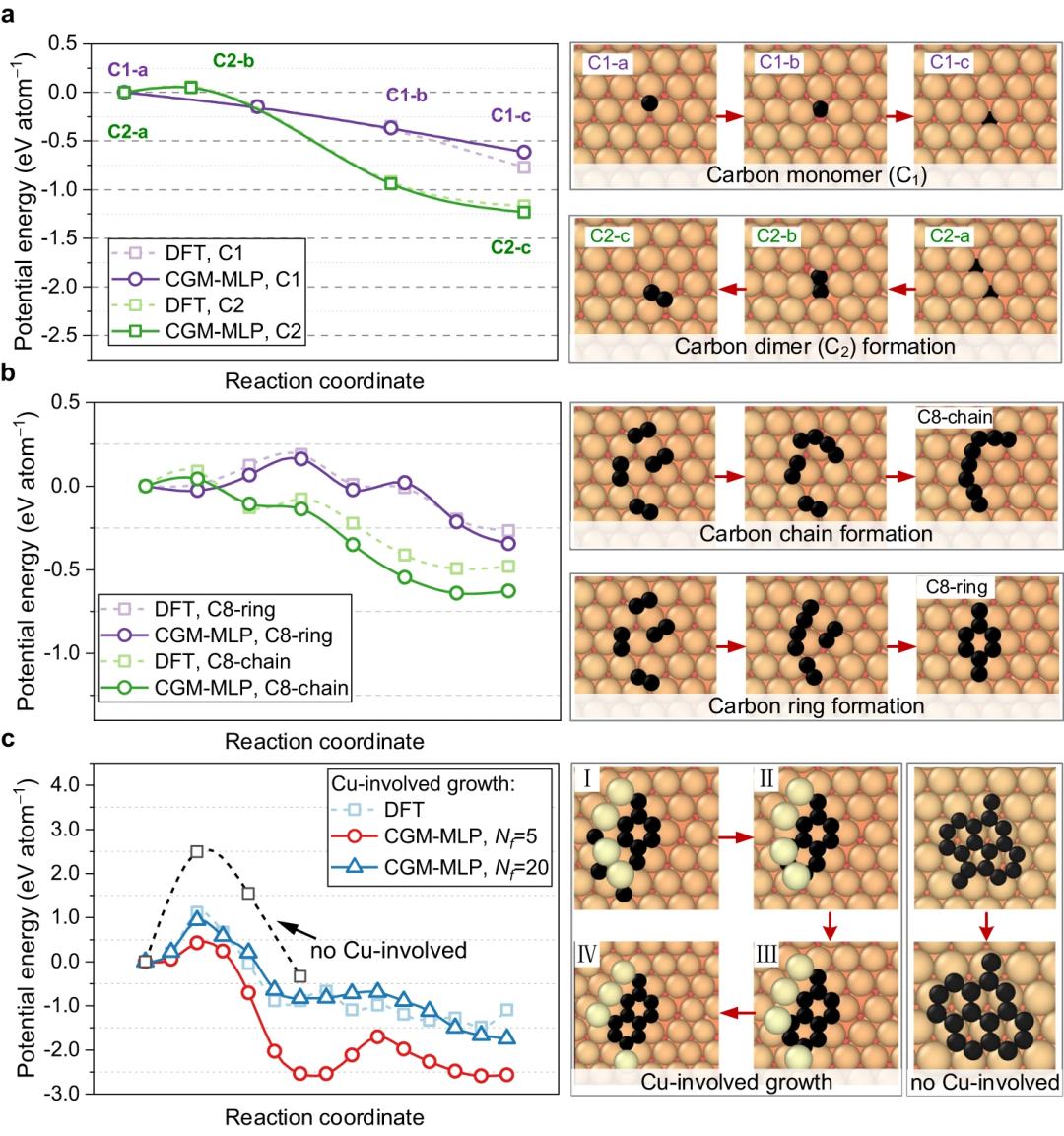
#Figure 4: Carbon diffusion and graphite obtained using CGM-MLP on metal and DFT-based crawling image nudging elastic band (CI-NEB) calculations The minimum energy path for ene nucleation. (Source: Paper)
Researchers investigated the initial nucleation of different metal surfaces, especially carbon deposition on Cu(111), Cr(110), Ti(001) and O-contaminated Cu(111) The simulations show consistency with experimental observations and DFT calculations.
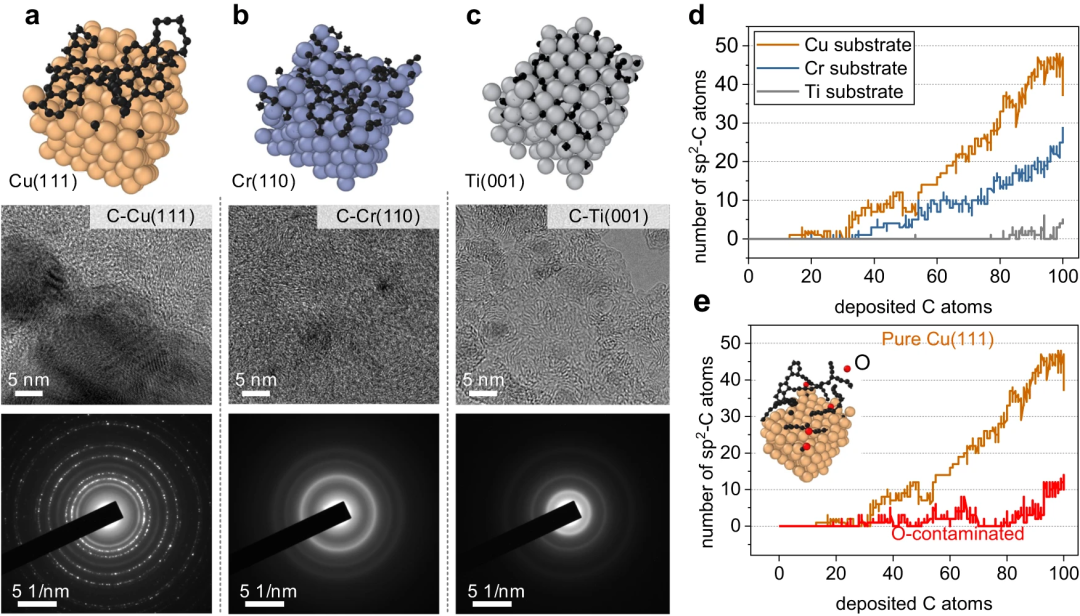
Figure 5: Representative metal surfaces for carbon nanostructure growth. (Source: Paper)
Research Significance
In summary, this research represents a pioneering advance in the integration of MLP and MD/tfMC for designing metal or alloy substrates provides a transferable and efficient strategy to obtain desired carbon nanostructures.
CGM-MLP effectively combines the accuracy of first principles methods with the efficiency of classical force fields. The tfMC method overcomes the time scale limitations of traditional AIMD or classical MD methods. Furthermore, the automated training framework of CGM-MLP incorporates specialized query strategies for building dynamic training sets in deposition simulations, emphasizing the importance of considering the local environment around deposited atoms.
These developments make it possible to directly study the mechanism of carbon growth on complex metal surfaces. The machine learning-driven deposition model proposed in this study may provide opportunities to study the growth of a variety of carbon nanostructures (e.g., graphene, carbon nanotubes, graphite, or diamond-like carbon films) on multi-element metal or alloy substrates.
The above is the detailed content of Shanghai Jiao Tong University team develops data-driven active learning framework to accelerate research progress in carbon nanomaterials. For more information, please follow other related articles on the PHP Chinese website!
















![[Web front-end] Node.js quick start](https://img.php.cn/upload/course/000/000/067/662b5d34ba7c0227.png)



