

Editor | Violet
The chemical space of synthesized molecules is very Vast. Effectively exploring this field requires relying on computational screening techniques, such as deep learning, to quickly discover a variety of interesting compounds.
Convert molecular structures into digital representations and develop corresponding algorithms to generate new Molecular structure is the key to chemical discovery.
Recently, a research team from the University of Glasgow in the UK proposed a machine learning model based on electron density training for generating host-guest binders. This model can simplify molecules The data is read in linear input specification (SMILES) format with an accuracy of up to 98%, thus achieving a comprehensive description of molecules in two-dimensional space.
Generates the electron density and electrostatic charge of the host-guest system through a variational autoencoder Three-dimensional representation of the potential, and then optimize the generation of guests through gradient descent. Finally, Transformer is used to convert the guests into SMILES, achieving effective representation and conversion of the guest structure.
The model is successfully applied to the established molecules The host system, cucurbituril and metal-organic cages, resulted in the discovery of 9 previously verified CB[6] guests and 7 unreported guests, and the discovery of 4 unreported 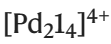 Object.
Object.
The research was titled "Electron density-based GPT for optimization and suggestion of host–guest binders" and was published in "Nature Computational Science" on March 8, 2024.

Current host-guest chemistry research is laborious and expensive
Strings, such as SMILES, molecules are represented by "words", such as "C1C=C1 ” (cyclopropene), is one of the most widespread numerical representations of molecules. Using state-of-the-art natural language processing, these representations are directly compatible with AI technologies such as recurrent neural networks or Transformer models.
The advantage of representing molecules as 3D volumes is that the latest AI technologies, such as convolutional neural networks, can be applied. To date, most applications of 3D volumes as molecular descriptors have focused on predicting properties or de novo drug design. However, the use of 3D volumes as molecular descriptors is currently hampered by the lack of efficient methods to relate these volumes to clear molecular structures.
Over the past 40 years, the main focus has been on molecular containers (hollow organic molecules or hollow supramolecular structures) that tend to alter the chemical and physical properties of molecules by isolating them from bulk phases in the cavity. Object systems have been increasingly studied. Host-guest systems have a wide range of applications, from catalysis to biomedical engineering, materials science, and stabilization of reactive molecules.
Cucurbituril (CB[n]) and metal-organic cages are among the most successful molecular container designs. Although host-guest chemistry has achieved remarkable achievements, the discovery of unreported guests in existing systems or the optimization of new host-guest systems remains a laborious and expensive iterative process that hinders the pace of scientific progress.
A machine learning model trained on electron density
Here, it is demonstrated that representing host molecules as 3D volumes (i.e., electrons modified with electrostatic potential Density) can be discovered through computer-assisted discovery of the host-guest system without knowledge of the host-guest system beyond the chemical structure of the host.
In the process, the researchers built a Transformer model that can be trained to efficiently convert 3D volumetric molecular descriptors into SMILES representations, thereby generating molecular structures usable by professional chemists.
The study also found that by modifying the molecule's electron density with electrostatic potential data, the molecule can be effectively represented as a 3D volume, and that these two features are sufficient to optimize the relationship between 3D descriptors by using an autoregressive sampling scheme. Volume shape and charge interact to discover the host's guest molecules.

The Transformer model perfectly predicts its SMILES representation with an accuracy of 98.125%. The prediction accuracy of a single token is 99.114%. Transformer's decoder Can also be isolated to purely generative models, such as GPT.
Workflow Overview
Computer-aided discovery of cucurbituril CB[6] and experimental validation of metal-organic cages  requires a two-tier workflow. First, an in silico workflow was designed to generate virtual libraries of potential guest molecules for both hosts. An in vitro workflow was then established that included the selection of the most promising guest candidates from these virtual libraries by expert chemists for experimental testing.
requires a two-tier workflow. First, an in silico workflow was designed to generate virtual libraries of potential guest molecules for both hosts. An in vitro workflow was then established that included the selection of the most promising guest candidates from these virtual libraries by expert chemists for experimental testing.
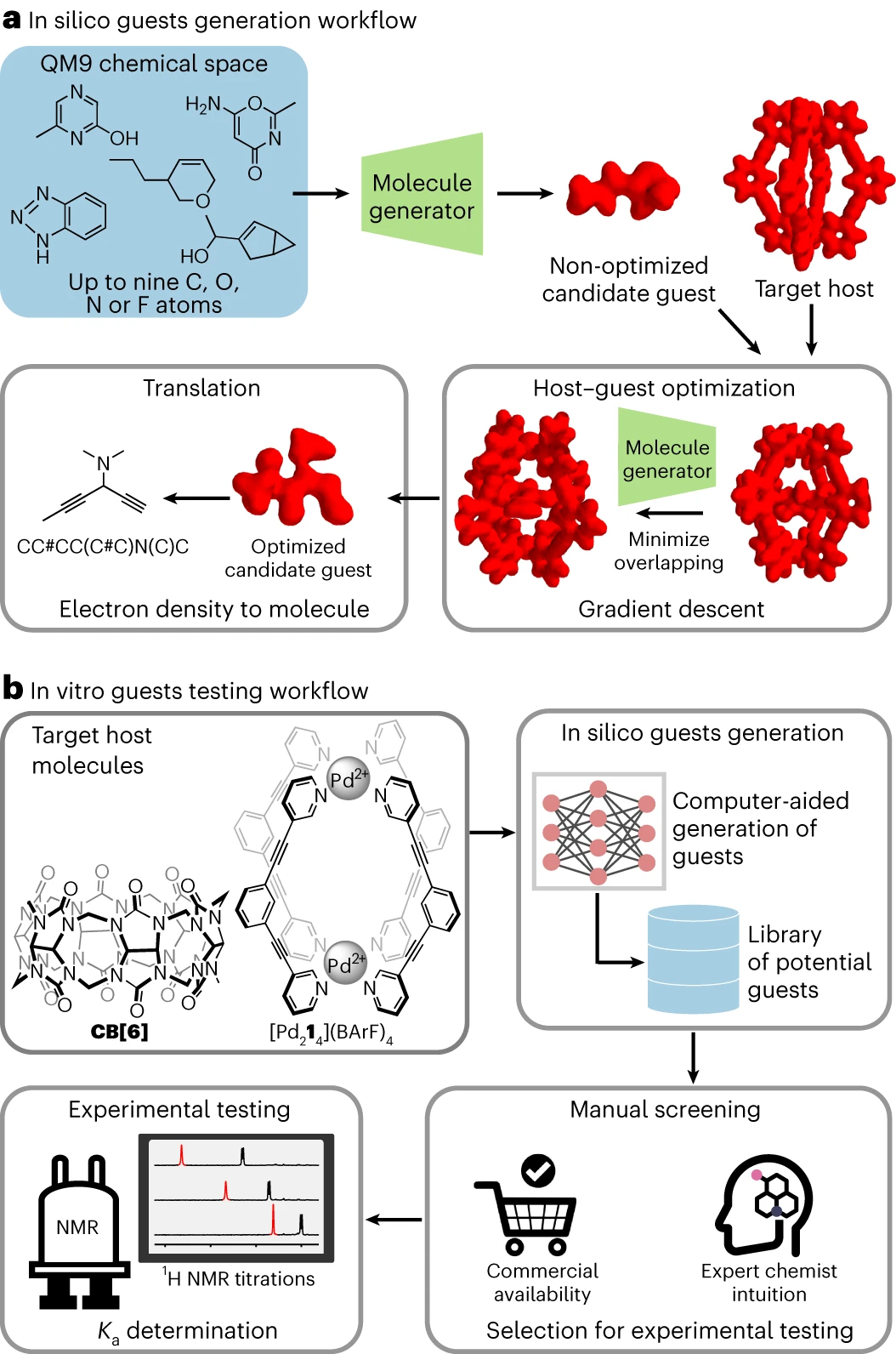
The computer generation of CB[6] and guest molecules is achieved through the workflow shown in the figure above. The workflow includes the following steps:
(1) The 3D electron density volume training set is derived from molecules in the publicly available QM9 dataset. Then, by modeling this 3D electron density volume training set using a variational autoencoder (VAE), a
"Molecular Generator", allowing the generation of 3D electron density volumes beyond those derived from the QM9 data set. The VAE molecule generator works by encoding a 3D electron density volume into a one-dimensional (1D) latent space and then generating the 3D electron density volume corresponding to the molecule by decoding from this 1D latent space. Interestingly, this approach only produced chemically sound molecules.
(2) The VAE molecule generator and gradient descent optimization algorithm are used to generate a library of guest molecules (in the form of 3D electron density volumes) for a given host molecule. Guest molecules are created by minimizing the overlap between host and guest electron densities while optimizing their electrostatic interactions.
(3) Since it can be challenging for human operators to convert 3D electron density volumes into chemically interpretable structures, the Transformer model was trained to convert these volumes into SMILES representations in a way that is more easily accessible to professionals. A format that chemists understand captures all the necessary information needed to describe a molecule. After generating potential guest molecules for CB[6] and through computer simulations, an in vitro workflow was established to experimentally test the most promising candidates.
The experimental procedures used are described below.
(1) The objects of CB[6] and generated due to its computer workflow are classified by chemical experts for experimental testing. Promising guests for testing are selected based on their structural similarity to known guests of CB[6] or , the intuition of professional chemists, and their commercial availability.
(2) Use direct 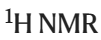 titration method to determine the affinity of CB[6] or . It is worth noting that, Guests generated in silico contain a mixture of molecules previously known to bind to (or be closely related to) the host and molecules that defy expert intuition.
titration method to determine the affinity of CB[6] or . It is worth noting that, Guests generated in silico contain a mixture of molecules previously known to bind to (or be closely related to) the host and molecules that defy expert intuition.
Experimental validation of two common host-guest systems
The researchers experimentally validated their workflow for two common host-guest systems: cucurbituril (CB[n]) and metal-organic cages, which became literature-validated and unreported guests.
The algorithm generated 9 previously known guests for CB[6]. It also identified 7 potential new guests for CB[6] that chemists deemed worthy of experimental testing. CB[6] The affinity of the new guest was assessed by direct  titration in HCO2H/H2O 1:1v/v.
titration in HCO2H/H2O 1:1v/v.
In all 7 cases, a set of signals for the host-guest system was observed, indicating that the system Rapid exchange occurs on the NMR time scale. After complexation, the aliphatic chain resonances of the guest molecules shift upfield, indicating that they are encapsulated within the CB[6] cavity. Found  The association constants with CB[6] follow previously established trends, ranging from 13.5 M^−1 to 5,470 M^−1.
The association constants with CB[6] follow previously established trends, ranging from 13.5 M^−1 to 5,470 M^−1.
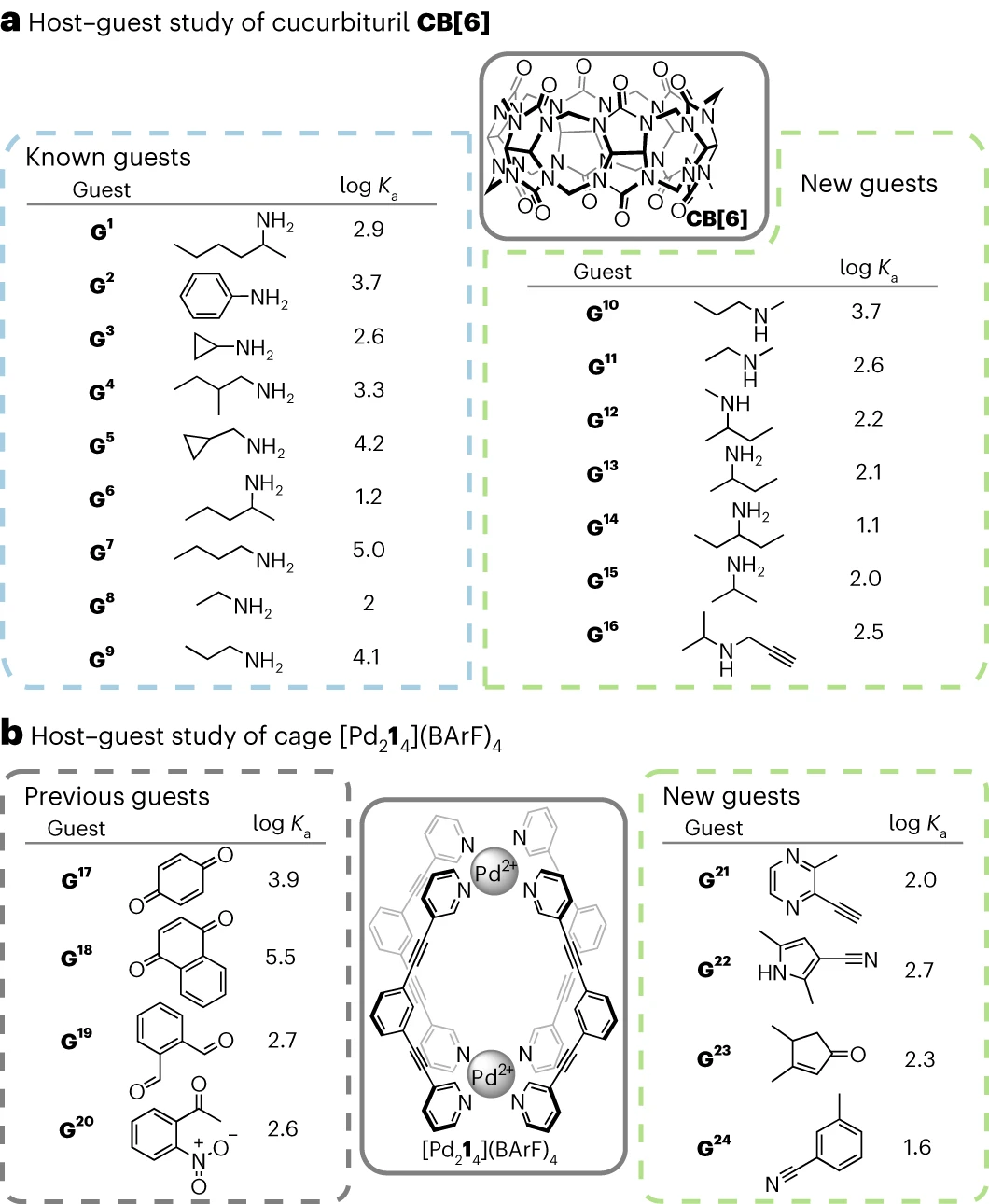
. (Source: paper) For , the optimization algorithm only generates unknown guest molecules, four potential unreported guests and [Pd214](BArF)4 The binding strength was tested by direct titration in CD2Cl2. In all four cases, the guest's affinity for [Pd214](BArF)4 was in the lower range of previously reported affinities for "small neutral guests" in CD2Cl2 Consistent (Ka from 44 M^-1 to 529 M^-1).
Although the QM9 data set contains perfectly sized molecules that can become guests of hosts such as CB[6], one limitation encountered by this study is that metal-organic cages have larger cavities and require larger object molecules. In future studies, datasets containing larger molecules, such as the GDB-17 dataset, will be used.
After that, “Our goal is to embed the selection of new ligands into the generation process, autonomously synthesize molecules on automated synthesis platforms (such as Chemputer robots), close the loop between optimization and testing, and create a Cyber-physical closed-loop system."
The above is the detailed content of Accuracy >98%, GPT based on electron density is used in chemical research, published in Nature sub-journal. For more information, please follow other related articles on the PHP Chinese website!
 How to solve the problem of missing ssleay32.dll
How to solve the problem of missing ssleay32.dll
 How to open the terminal window in vscode
How to open the terminal window in vscode
 Is A5 bigger or B5 paper bigger?
Is A5 bigger or B5 paper bigger?
 The role of parseint function
The role of parseint function
 The difference between arrow functions and ordinary functions
The difference between arrow functions and ordinary functions
 How to define an array
How to define an array
 How much is Snapdragon 8gen2 equivalent to Apple?
How much is Snapdragon 8gen2 equivalent to Apple?
 fil currency price real-time price
fil currency price real-time price
 SpringBoot project building steps
SpringBoot project building steps








![[Web front-end] Node.js quick start](https://img.php.cn/upload/course/000/000/067/662b5d34ba7c0227.png)



