


Des équipes de recherche de l'Université Tsinghua AIR, de l'Université de Pékin et de l'Université de Nanjing ont proposé le modèle ESM-AA. Ce modèle a réalisé des progrès importants dans le domaine de la modélisation du langage protéique, fournissant une solution de modélisation unifiée intégrant des informations multi-échelles.
Il s'agit du premier modèle de langage protéique pré-entraîné capable de gérer à la fois les informations sur les acides aminés et les informations atomiques. Les excellentes performances du modèle démontrent le grand potentiel de la modélisation unifiée multi-échelle pour surmonter les limitations existantes et débloquer de nouvelles capacités.
En tant que modèle de base, ESM-AA a reçu l'attention et des discussions approfondies de la part de nombreux chercheurs (voir capture d'écran ci-dessous) et est considéré comme ayant le potentiel de développer des modèles basés sur ESM-AA qui peuvent rivaliser avec AlphaFold3 et RoseTTAFold All-Atom. Cela ouvre de nouvelles voies pour étudier les interactions entre différentes structures biologiques. Le document actuel a été accepté par l'ICML 2024.
Les protéines sont l'exécuteur clé de diverses activités de la vie. La compréhension approfondie des protéines et de leurs interactions avec d’autres structures biologiques est une question centrale en sciences biologiques, qui revêt une importance pratique significative pour le criblage ciblé de médicaments, l’ingénierie enzymatique et d’autres domaines.
Par conséquent, comment mieux comprendre et modéliser les protéines est devenu un point chaud de la recherche dans le domaine de l'AI4Science.
Ces derniers jours, de grandes institutions de recherche de pointe, dont Deepmind et le Baker Group de l'Université de Washington, ont également mené des recherches approfondies sur le problème de la modélisation des protéines par tous les atomes et ont proposé des méthodes telles que AlphaFold 3, RoseTTAFold. All-Atom, etc. pour les protéines et d'autres activités de la vie. Le modèle de modélisation à l'échelle entièrement atomique des molécules associées peut réaliser des prédictions précises de la structure des protéines, de la structure moléculaire, de la structure du récepteur-ligand et d'autres échelles entièrement atomiques avec une grande précision.
Bien que ces modèles aient fait des progrès significatifs dans la modélisation structurelle à l’échelle entièrement atomique, les modèles actuels de langage protéique grand public sont toujours incapables de parvenir à la compréhension et à l’apprentissage de la représentation des protéines à l’échelle entièrement atomique.
Les modèles d'apprentissage de la représentation protéique représentés par ESM-2 utilisent les acides aminés comme seule échelle pour construire des modèles, ce qui convient aux situations qui se concentrent sur la transformation des protéines est une approche raisonnable.
Cependant, la clé pour bien comprendre la nature des protéines réside dans l'élucidation de leurs interactions avec d'autres structures biologiques telles que les petites molécules, l'ADN, l'ARN, etc.
Face à cette demande, il est nécessaire de décrire les interactions complexes entre différentes structures, et il est difficile pour une stratégie de modélisation à échelle unique de fournir une couverture complète et efficace.
Pour pallier cette lacune, les modèles protéiques connaissent une profonde innovation vers des modèles multi-échelles. Par exemple, le modèle RoseTTAFold All-Atom publié dans le magazine Science début mai, en tant que produit de suivi de RoseTTAFold, a introduit le concept de multi-échelle.
Ce modèle ne se limite pas à la prédiction de la structure des protéines, mais s'étend également à des domaines de recherche plus larges tels que l'amarrage des protéines et des molécules/acides nucléiques, les modifications post-traductionnelles des protéines, etc.
Dans le même temps, AlphaFold3, récemment publié par DeepMind, adopte également une stratégie de modélisation multi-échelle pour prendre en charge la prédiction de la structure de plusieurs complexes protéiques. Ses performances sont impressionnantes et auront sans aucun doute un impact majeur sur les domaines de l'intelligence artificielle et de la biologie. .
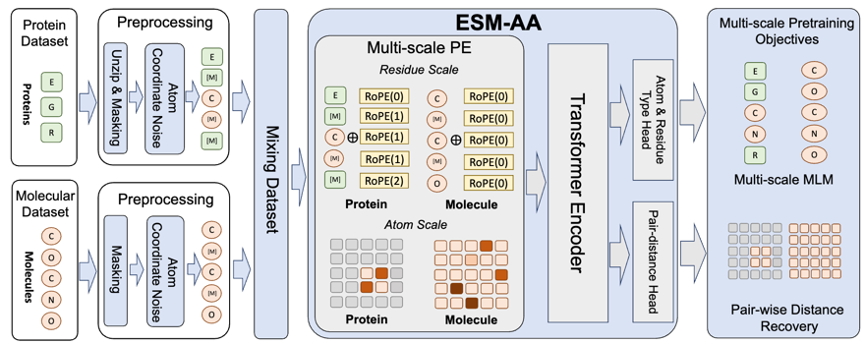
L'application réussie de RoseTTAFold All-Atom et AlphaFold3 au multi- Le concept d'échelle a inspiré une réflexion importante : comment le modèle de langage protéique en tant que modèle de base protéique devrait adopter une technologie multi-échelle. Sur cette base, l’équipe a proposé le modèle de langage protéique multi-échelle ESM All-Atom (ESM-AA).
En bref, ESM-AA introduit le concept de multi-échelle en « décompressant » certains acides aminés dans la composition atomique correspondante. Par la suite, un pré-entraînement a été réalisé en mélangeant des données protéiques et des données moléculaires, ce qui a donné au modèle la capacité de gérer simultanément des structures biologiques de différentes échelles.
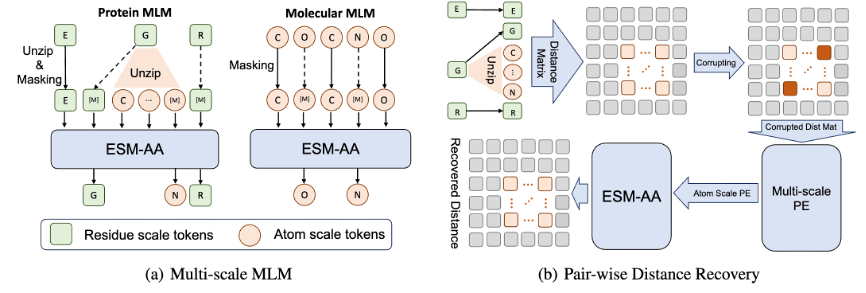
De plus, afin d'aider le modèle à mieux apprendre des informations de haute qualité à l'échelle atomique, ESM-AA utilisera également des données de structure moléculaire à l'échelle atomique pour la formation. De plus, en introduisant le mécanisme de codage de position multi-échelle illustré à la figure 2, le modèle ESM-AA peut bien distinguer les informations à différentes échelles, garantissant ainsi que le modèle peut comprendre avec précision les informations de position et de structure au niveau des résidus et au niveau atomique.
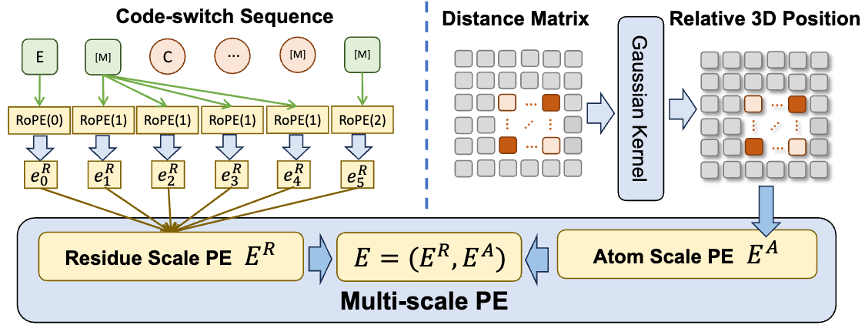
Pour aider le modèle à apprendre des informations multi-échelles, l'équipe a conçu une variété d'objectifs de pré-formation pour le modèle ESM-AA. Les objectifs de pré-formation multi-échelles de l'ESM-AA incluent la modélisation du langage masqué (MLM) et la récupération à distance par paire (PDR). Comme le montre la figure 3 (a), MLM nécessite que le modèle fasse des prédictions basées sur le contexte environnant en masquant les acides aminés et les atomes. Cette tâche de formation peut être effectuée à la fois aux échelles des acides aminés et des atomes. Le PDR nécessite que le modèle prédise avec précision la distance euclidienne entre différents atomes afin d'entraîner le modèle à comprendre les informations structurelles au niveau atomique (comme le montre la figure 3 (b)).
Le modèle ESM-AA est affiné et évalué sur plusieurs tâches de référence de protéines et de petites molécules, y compris les enzymes -tâche de régression d'affinité de substance de substrat (les résultats sont présentés dans la figure 4), tâche de classification de paires enzyme-substrat (les résultats sont présentés dans la figure 4) et tâche de régression d'affinité médicament-cible (les résultats sont présentés dans la figure 5).
Les résultats montrent qu'ESM-AA surpasse les modèles précédents dans ces tâches, indiquant qu'il réalise pleinement le potentiel des modèles de langage protéiques pré-entraînés aux échelles des acides aminés et des atomes.
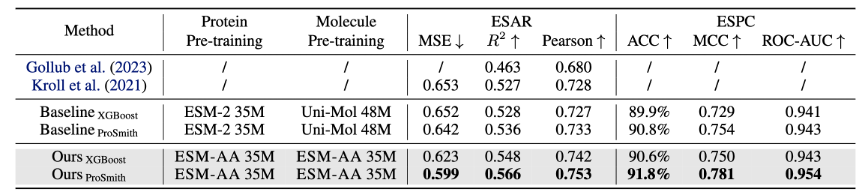
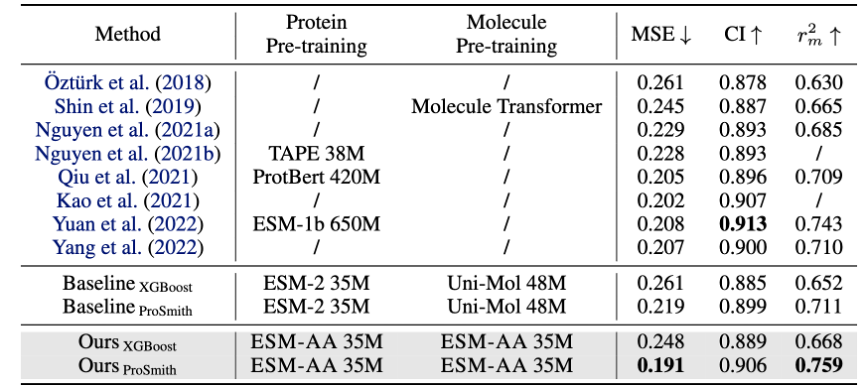
Figure 5 : Comparaison des performances de la tâche de régression par affinité médicament-cible
De plus, l'ESM-AA modèle également Les performances ont été testées sur des tâches telles que la prédiction du contact des protéines, la classification fonctionnelle des protéines et la prédiction des propriétés moléculaires.
Les résultats montrent que lorsqu'il s'agit de tâches impliquant uniquement des protéines, ESM-AA fonctionne à égalité avec ESM-2 sur les tâches moléculaires, le modèle ESM-AA surpasse la plupart des modèles de référence et est similaire à Uni-Mol.
Cela montre que l'ESM-AA ne sacrifie pas sa capacité à comprendre les protéines dans le processus d'acquisition de connaissances moléculaires puissantes. Cela illustre également que le modèle ESM-AA réutilise avec succès les connaissances du modèle ESM-2 sans avoir à repartir de là. développement scratch, réduisant considérablement les coûts de formation des modèles.
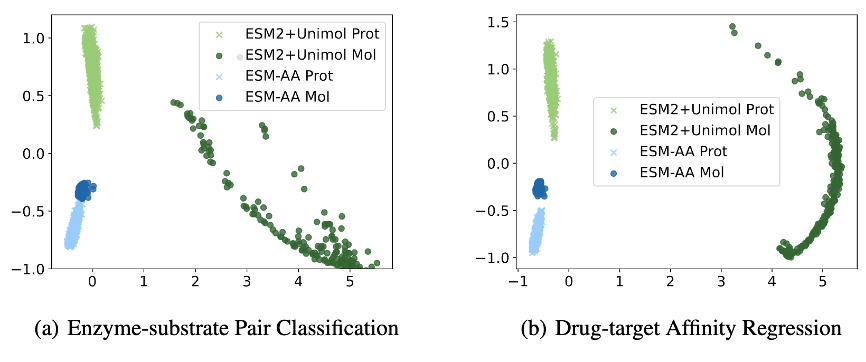
Afin d'analyser plus en détail les raisons pour lesquelles ESM-AA fonctionne bien sur la tâche de référence protéine-petite molécule, cet article montre l'extraction du modèle ESM-AA et de l'ESM-2+Uni-Mol. combinaison de modèles dans cette tâche Visualisation de la distribution de la représentation de l'échantillon.
Comme le montre la figure 6, les représentations de protéines et de petites molécules apprises par le modèle ESM-AA sont plus compactes, ce qui indique que les deux sont dans le même espace de représentation. C'est pourquoi le modèle ESM-AA est meilleur que l'ESM. -2+Uni- Modèle Mol, illustrant davantage les avantages de la modélisation moléculaire unifiée multi-échelle.
Conclusion
L'ESM-AA développé par l'équipe Tsinghua AIR est le premier modèle de langage protéique pré-entraîné qui intègre le traitement des acides aminés et de l'information atomique. Le modèle démontre des performances robustes et excellentes en intégrant des informations multi-échelles, offrant ainsi une nouvelle façon de résoudre le problème des interactions entre les structures biologiques.
ESM-AA favorise non seulement une compréhension plus approfondie des protéines, mais fonctionne également bien dans plusieurs tâches biomoléculaires, prouvant qu'il peut intégrer efficacement les connaissances au niveau moléculaire tout en maintenant les capacités de compréhension des protéines, réduisant ainsi le coût de la formation des modèles. nouvelle direction pour la recherche biologique assistée par l’IA.
Titre de l'article : ESM All-Atom : Modèle de langage protéique multi-échelle pour la modélisation moléculaire unifiée
Ce qui précède est le contenu détaillé de. pour plus d'informations, suivez d'autres articles connexes sur le site Web de PHP en chinois!
 Comment flasher le téléphone Xiaomi
Comment flasher le téléphone Xiaomi
 Comment centrer un div en CSS
Comment centrer un div en CSS
 Comment ouvrir un fichier rar
Comment ouvrir un fichier rar
 Méthodes de lecture et d'écriture de fichiers Java DBF
Méthodes de lecture et d'écriture de fichiers Java DBF
 Comment résoudre le problème de l'absence du fichier msxml6.dll
Comment résoudre le problème de l'absence du fichier msxml6.dll
 Formules de permutation et de combinaison couramment utilisées
Formules de permutation et de combinaison couramment utilisées
 Numéro de téléphone mobile virtuel pour recevoir le code de vérification
Numéro de téléphone mobile virtuel pour recevoir le code de vérification
 album photo dynamique
album photo dynamique








![[Web front-end] Démarrage rapide de Node.js](https://img.php.cn/upload/course/000/000/067/662b5d34ba7c0227.png)



