


La dynamique d'une protéine est cruciale pour comprendre son mécanisme. Cependant, la prévision informatique des informations cinétiques des protéines est un défi.
Ici, une équipe de recherche de l'Université du Shandong, de BioMap, de l'Institut de technologie de Pékin, du Hubei Medical College, de l'Université médicale de Ningxia et de l'Université des sciences et technologies King Abdullah (KAUST) a proposé un modèle de réseau neuronal RMSF-net, qui surpasse les méthodes précédentes et produit les meilleurs résultats sur des ensembles de données sur la dynamique des protéines à grande échelle ; le modèle peut déduire avec précision les informations dynamiques d’une protéine en quelques secondes.
En apprenant efficacement de l'intégration de données expérimentales sur la structure des protéines et de données cryo-EM, cette méthode est capable d'identifier avec précision les contraintes bidirectionnelles interactives et la supervision entre les images cryo-EM et les modèles PDB pour maximiser l'efficacité de la prédiction dynamique.
RMSF-net est un outil gratuit qui jouera un rôle important dans les études sur la dynamique des protéines.
L'étude s'intitulait « Prédiction précise de la flexibilité structurelle des protéines par l'apprentissage profond intégrant des structures atomiques complexes et des informations sur la densité cryo-EM » et a été publiée dans « Nature Communications » le 2 juillet.

Adresse GitHub RMSF-net :
Dynamique des protéines
La dynamique des protéines est cruciale pour comprendre leurs mécanismes. La technologie de microscopie cryoélectronique (cryo-EM) peut résoudre la plupart des protéines, où la structure macromoléculaire est représentée par une carte de densité 3D.
Limitations de la cryomicroscopie électronique
En raison de la faible résolution et du rapport signal/bruit des images de particules 2D originales, l'analyse par cryomicroscopie électronique ne peut pas résoudre de petits changements conformationnels pendant le processus de reconstruction.
Application du deep learning en cryomicroscopie électronique
Les méthodes d'apprentissage profond sont largement utilisées dans l'analyse automatique des images de cryomicroscopie électronique. À l’aide de cartes cryo-EM haute résolution, un modèle de banque de données sur les protéines (PDB) peut être construit à partir des cartes cryo-EM.
Présentation de RMSF-net
RMSF-net est un modèle de réseau neuronal pour les cartes de densité de cryomicroscopie électronique. Il exploite la densité cryo-EM et les informations du modèle PDB pour déduire avec précision les informations dynamiques des protéines en quelques secondes.
RMSF
RMSF est une méthode de mesure largement utilisée pour évaluer la flexibilité des structures moléculaires dans les analyses de dynamique moléculaire (MD). Son objectif principal est de prédire le RMSF des structures locales (résidus, atomes) au sein d'une protéine.
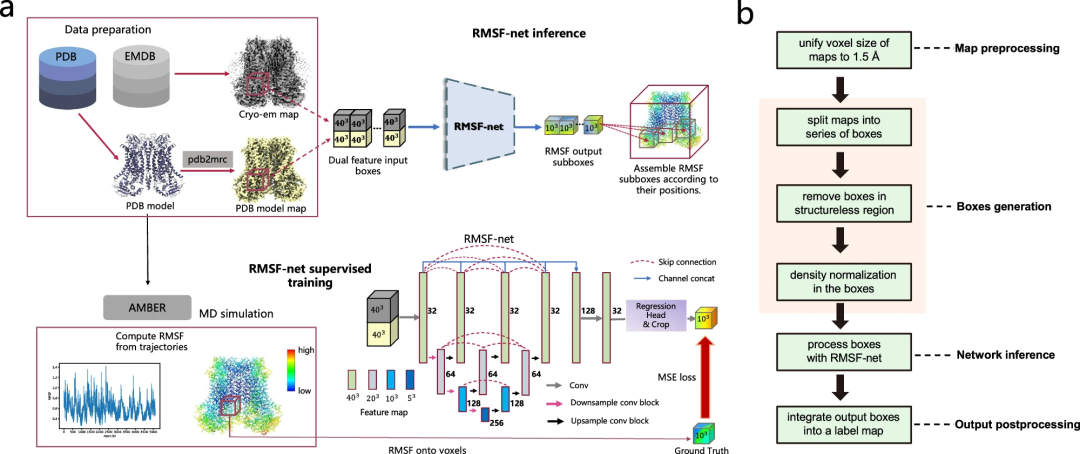
En plus des images cryo-EM, RMSF-net utilise des modèles PDB comme entrée supplémentaire pour produire des prédictions RMSF très proches des résultats de simulation MD.
RMSF-net est un réseau neuronal convolutif tridimensionnel contenant deux modules interconnectés. Le module principal utilise l'architecture Unet+ (L3) pour encoder et décoder les fonctionnalités des boîtes de densité d'entrée. Un autre module utilise des convolutions 1x1 pour régresser les canaux des cartes de fonctionnalités générées par le backbone Unet+. Le découpage central est ensuite appliqué à la sortie du module de régression pour obtenir une sous-boîte RMSF centrée, où la valeur du voxel correspond au RMSF des atomes qu'elle contient. Enfin, les sous-boîtes RMSF sont fusionnées spatialement dans une carte RMSF à l'aide d'un algorithme de fusion.
En outre, les chercheurs ont également construit un ensemble de données à grande échelle sur la dynamique des protéines pour la formation et la validation de RMSF-net, dans lequel 335 entrées structurelles cryo-EM avec des modèles PDB adaptés ont été sélectionnées et des simulations MD correspondantes ont été effectuées. Des résultats expérimentaux complets démontrent l’efficience et l’efficacité de RMSF-net.
Tableau : Performances des différentes méthodes de prédiction RMSF sur l'ensemble de données. (Source : article)
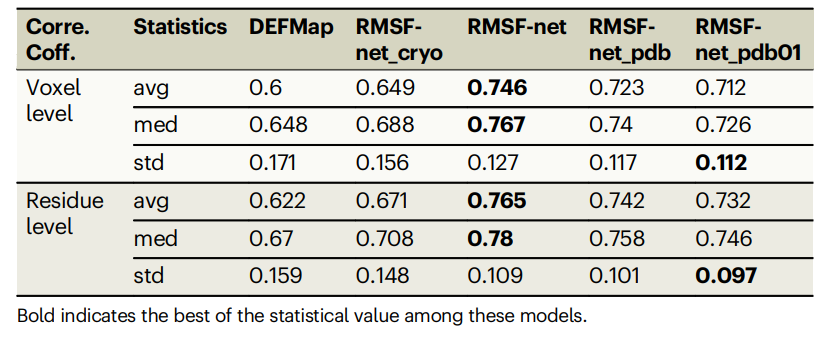
RMSF-net a obtenu de bons résultats lors d'une validation croisée rigoureuse de 5 fois, avec un coefficient de corrélation de 0,746 ± 0,127 avec les résultats de simulation MD. Le coefficient de corrélation de RMSF-net est amélioré de 15 % par rapport à DEFMap et de 10 % par rapport à la méthode de base.
Interprétabilité des prédictions dynamiques
Les chercheurs ont amélioré l'interprétabilité des prédictions dynamiques RMSF-net grâce à des expériences comparatives. Ils divisent le processus de prévision RMSF en deux étapes :
Des études ont montré que la prédiction dynamique des modèles basés sur des spectres de cryo-microscopie électronique (tels que DEFMap ou RMSF-net_cryo) se fait principalement par interprétation des protéines Implémentation de la structure. Cela met en évidence le lien entre la topologie et la dynamique des protéines, conformément aux premiers principes des relations structure-fonction.
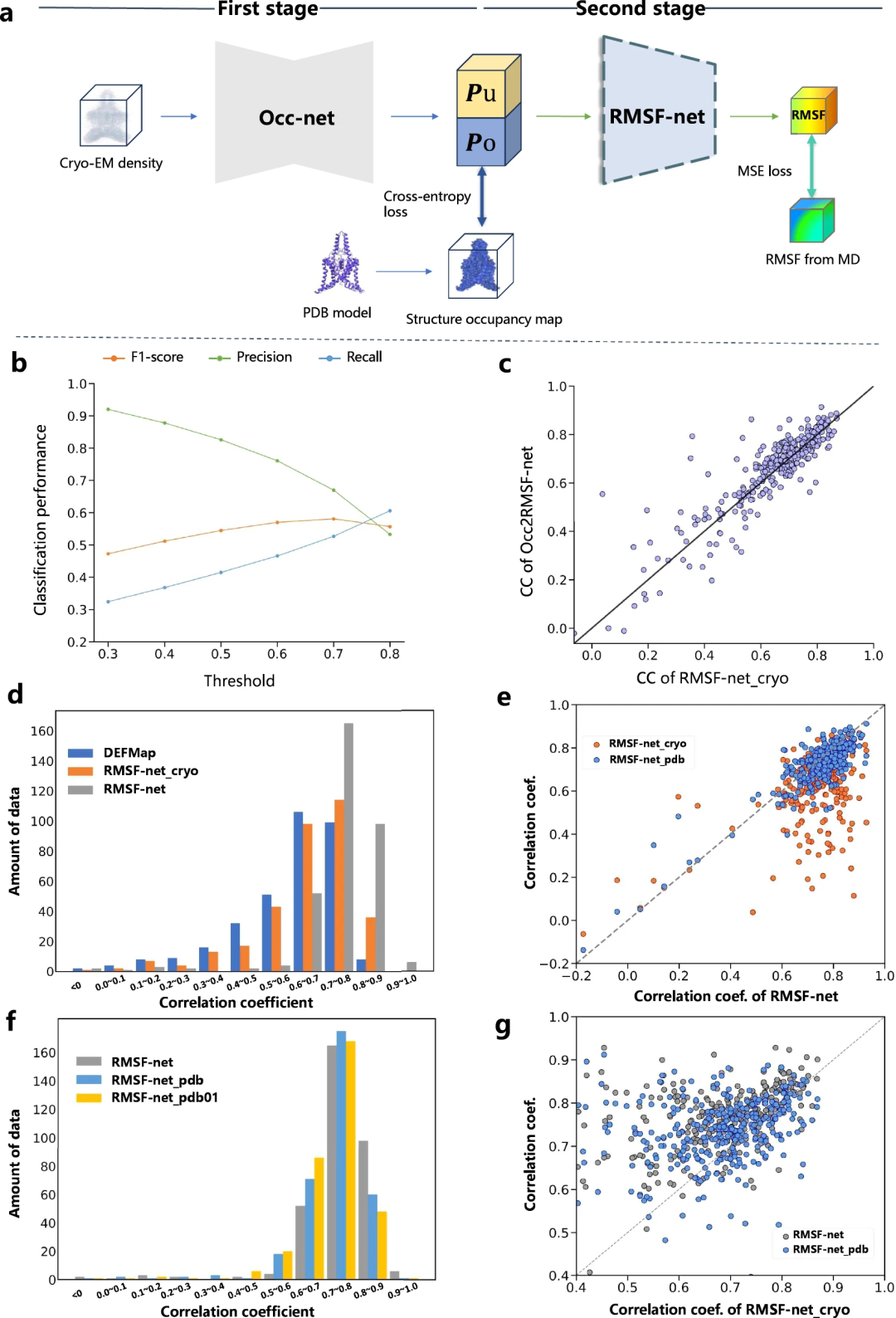
De plus, grâce à une comparaison complète de RMSF-net_cryo, RMSF-net_pdb et de la double combinaison finale RMSF-net, il est prouvé que : d'une part, les informations structurelles du modèle PDB jouent un rôle majeur dans le rôle RMSF-net, où le modèle profond apprend des modèles entre la topologie structurelle et la flexibilité à partir de simulations MD, et d'autre part, le modèle est encore amélioré par les informations cinétiques contenues dans la distribution de densité hétérogène de la cryomicroscopie électronique Plans. Ces résultats valident le rôle complémentaire des informations provenant des cartes cryo-EM et des modèles PDB sur la prédiction de la dynamique des protéines dans RMSF-net.
Limitations et orientations futures
Il est indéniable que RMSF-net se limite principalement à prédire la flexibilité des protéines pures et de leurs complexes en solution. Cette méthode peut présenter des inexactitudes dans certaines régions localisées concernant les propriétés dynamiques de la protéine lorsqu’elle est liée à des ligands de petites molécules ou dans un environnement membranaire.
Les excellentes performances de RMSF-net révèlent la faisabilité de recherches plus approfondies dans cette direction. Ces recherches n'ont pas été étendues aux acides nucléiques et aux complexes protéine-acide nucléique. La caractérisation complète de tous les aspects de la dynamique macromoléculaire, y compris la prédiction multiconformationnelle et l'analyse des transitions, nécessite des recherches plus approfondies et approfondies à l'avenir.
Néanmoins, en tant qu'outil de prédiction de la dynamique des protéines, RMSF-net a encore de grandes perspectives d'application dans la recherche sur la structure et la dynamique des protéines en raison de ses performances supérieures et de sa vitesse de traitement ultra-rapide.
Remarque : la couverture provient d'Internet
Ce qui précède est le contenu détaillé de. pour plus d'informations, suivez d'autres articles connexes sur le site Web de PHP en chinois!
 Comment résoudre 404 introuvable
Comment résoudre 404 introuvable
 Causes et solutions des erreurs d'exécution
Causes et solutions des erreurs d'exécution
 Méthode de mise en œuvre de la fonction de lecture vocale Android
Méthode de mise en œuvre de la fonction de lecture vocale Android
 que signifie pm
que signifie pm
 La différence entre xdata et data
La différence entre xdata et data
 Comment vérifier l'utilisation du processeur sous Linux
Comment vérifier l'utilisation du processeur sous Linux
 Comment utiliser onclick en HTML
Comment utiliser onclick en HTML
 puissance de l'ordinateur portable
puissance de l'ordinateur portable








![[Web front-end] Démarrage rapide de Node.js](https://img.php.cn/upload/course/000/000/067/662b5d34ba7c0227.png)



