
 Périphériques technologiques
IA
Plus rapide que 0 ! Meta a lancé un grand modèle protéique avec 15 milliards de paramètres pour écraser AlphaFold2
Périphériques technologiques
IA
Plus rapide que 0 ! Meta a lancé un grand modèle protéique avec 15 milliards de paramètres pour écraser AlphaFold2
Plus rapide que 0 ! Meta a lancé un grand modèle protéique avec 15 milliards de paramètres pour écraser AlphaFold2

Le plus grand modèle de langage protéique à ce jour a été publié !
Il y a un an, AlphaFold2, l’open source de DeepMind, a été lancé dans Nature et Science, bouleversant les cercles universitaires de la biologie et de l’IA.
Un an plus tard, Meta est arrivé avec ESMFold, qui était un ordre de grandeur plus rapide.
Non seulement il est rapide, mais le modèle comporte également 15 milliards de paramètres.

LeCun a tweeté pour faire l'éloge, il s'agit d'une belle nouvelle réalisation de l'équipe protéique Meta-FAIR.

Le co-auteur Zeming Lin a révélé que le grand modèle avec 3 milliards de paramètres a été formé sur 256 GPU pendant 3 semaines, tandis qu'ESMfold a pris 10 jours sur 128 GPU. Quant à la version à 15 milliards de paramètres, elle n'est toujours pas claire.
Il a également dit que le code sera définitivement open source plus tard, alors restez à l'écoute !
Grand et rapide !
Aujourd'hui, notre protagoniste est ESMFold, un modèle qui prédit directement la structure de haute précision, de bout en bout, au niveau atomique à partir de séquences protéiques individuelles.

Adresse papier : https://www.biorxiv.org/content/10.1101/2022.07.20.500902v1
Inutile de dire les avantages apportés par 15 milliards de paramètres - grâce à la formation, les grands modèles d'aujourd'hui peuvent prédire les trois -structure dimensionnelle des protéines avec une précision de taille atomique.
En termes de précision, ESMFold est presque le même qu'AlphaFold2 et RoseTTAFold.
Cependant, la vitesse d'inférence d'ESMFold est d'un ordre de grandeur plus rapide que celle d'AlphaFold2 !
Il peut être difficile de comprendre la comparaison de vitesse entre les trois en parlant d'ordre de grandeur. Il suffit de regarder l'image ci-dessous pour comprendre.

Quelle est la différence ?
Bien qu'AlphaFold2 et RoseTTAFold aient obtenu un succès révolutionnaire sur le problème de la prédiction de la structure par résolution atomique, ils s'appuient également sur l'utilisation d'alignements de séquences multiples (MSA) et de modèles de structure protéique similaires pour obtenir des performances optimales.
En revanche, en tirant parti de la représentation interne du modèle de langage, ESMFold peut générer des prédictions de structure correspondantes en utilisant une seule séquence comme entrée, accélérant ainsi considérablement la prédiction de structure.

Les chercheurs ont découvert que les prédictions d’ESMFold pour les séquences de faible complexité étaient comparables aux modèles de pointe actuels.
De plus, la précision de la prédiction de la structure est étroitement liée à la complexité du modèle de langage, c'est-à-dire que lorsque le modèle de langage peut mieux comprendre la séquence, il peut mieux comprendre la structure.

Actuellement, il existe des milliards de séquences protéiques de structure et de fonction inconnues, dont beaucoup sont dérivées du séquençage métagénomique.
Grâce à ESMFold, les chercheurs peuvent plier un échantillon aléatoire de 1 million de séquences métagénomiques en seulement 6 heures.

Une grande partie d'entre eux ont un niveau de confiance élevé et ne ressemblent à aucune structure connue (n'ont aucun enregistrement dans la base de données).
Les chercheurs pensent qu’ESMFold peut aider à comprendre les structures protéiques qui dépassent la compréhension actuelle.

De plus, comme les prédictions d'ESMFold sont d'un ordre de grandeur plus rapides que les modèles existants, les chercheurs peuvent utiliser ESMFold pour aider à combler le fossé entre les bases de données de séquences protéiques à croissance rapide et les bases de données sur la structure et la fonction des protéines à croissance plus lente.
Modèle de langage protéique à 15 milliards de paramètres
Parlons ensuite en détail du nouvel ESMFold de Meta.
ESM-2 est un modèle de langage basé sur Transformer et utilise un mécanisme d'attention pour apprendre les modèles d'interaction entre les paires d'acides aminés dans la séquence d'entrée.
Par rapport au modèle de génération précédente ESM-1b, Meta a amélioré la structure du modèle et les paramètres de formation, et ajouté des ressources et des données informatiques. Dans le même temps, l’ajout de l’intégration de positions relatives permet de généraliser le modèle à des séquences de n’importe quelle longueur.
D'après les résultats, le modèle ESM-2 avec 150 millions de paramètres a mieux fonctionné que le modèle ESM-1b avec 650 millions de paramètres.
De plus, ESM-2 surpasse également les autres modèles de langage protéique en termes de prédiction de structure. Cette amélioration des performances est cohérente avec les modèles établis dans le domaine de la modélisation linguistique à grande échelle.

À mesure que l'échelle d'ESM-2 augmente, une grande amélioration de la précision de la modélisation du langage peut être observée.

Prédiction de structure de séquence unique de bout en bout
Une différence clé entre SMFold et AlphaFold2 est que ESMFold utilise la représentation de modèle de langage, éliminant le besoin de séquences homologues explicites (sous forme de MSA) en entrée.
ESMFold simplifie l'Evoformer dans AlphaFold2 en remplaçant le module réseau coûteux en termes de calcul qui gère MSA par un module Transformer qui gère les séquences. Cette simplification signifie qu'ESMFold est nettement plus rapide que les modèles basés sur MSA.
La sortie du squelette replié est ensuite traitée par un module de structure, qui est responsable de la sortie de la structure finale au niveau atomique et de la confiance des prédictions.

Les chercheurs ont comparé ESMFold avec AlphaFold2 et RoseTTAFold sur les ensembles de tests CAMEO (avril 2022 à juin 2022) et CASP14 (mai 2020).
Lorsqu'une seule séquence est donnée en entrée, ESMFold fonctionne bien mieux qu'Alphafold 2.
Et en utilisant le pipeline complet, AlphaFold2 a obtenu respectivement 88,3 et 84,7 sur CAMEO et CASP14. ESMFold atteint une précision comparable à RoseTTAfold sur CAMEO, avec un score TM moyen de 82,0.

Conclusion
Les chercheurs ont découvert qu'un modèle de langage ciblant l'apprentissage non supervisé, formé sur une vaste base de données de séquences protéiques évolutivement diverses, était capable de prédire la structure des protéines à une résolution au niveau atomique.
En élargissant les paramètres du modèle de langage à 15B, l'impact de l'échelle sur l'apprentissage de la structure des protéines peut être systématiquement étudié.
Nous voyons que la courbe non linéaire des prédictions de la structure des protéines est fonction de la taille du modèle et observons un lien étroit entre la façon dont un modèle de langage comprend une séquence et ses prédictions de structure.
Les modèles de la série ESM-2 sont les plus grands modèles de langage protéique formés à ce jour, avec seulement un ordre de grandeur en moins de paramètres que les plus grands modèles de texte récemment développés.
De plus, ESM-2 constitue une très grande amélioration par rapport au modèle précédent, même à 150 millions de paramètres, ESM-2 capture une carte structurelle plus précise que le modèle de langage de la génération ESM-1 à 650 millions de paramètres.
Les chercheurs ont déclaré que le principal moteur des performances d'ESMFold est le modèle de langage. Parce qu’il existe un lien étroit entre la perplexité des modèles de langage et l’exactitude des prédictions de structure, ils ont découvert que lorsque l’ESM-2 peut mieux comprendre les séquences protéiques, il peut réaliser des prédictions comparables aux modèles de pointe actuels.
ESMFold a obtenu une prédiction précise de la structure de résolution atomique, et le temps d'inférence est d'un ordre de grandeur plus rapide qu'AlphaFold2.
En pratique, l’avantage de vitesse est encore plus grand. Parce qu'ESMFold n'a pas besoin de rechercher des séquences liées à l'évolution pour construire le MSA.
Bien qu'il existe des moyens plus rapides de réduire le temps de recherche, celui-ci peut rester très long, quelle que soit la manière dont vous le réduisez.
Les avantages apportés par le temps d'inférence considérablement réduit sont évidents : l'augmentation de la vitesse permettra de cartographier l'espace structurel de grandes bases de données de séquences métagénomiques.
En plus des outils basés sur la structure pour identifier l'homologie et la conservation distales, la prédiction rapide et précise de la structure avec ESMFold peut également jouer un rôle important dans l'analyse structurelle et fonctionnelle de grandes nouvelles collections de séquences.
L'accès à des millions de structures prédites dans un temps limité est propice à la découverte de nouvelles connaissances sur l'étendue et la diversité des protéines naturelles et permet la découverte de structures et de fonctions protéiques complètement nouvelles.
Introduction à l'auteur
Le co-auteur de cet article est Zeming Lin de Meta AI.

Selon sa page d'accueil personnelle, Zeming a étudié pour un doctorat à l'Université de New York et a travaillé comme ingénieur de recherche (visiteur) chez Meta AI, principalement responsable des travaux d'infrastructure back-end.
Il a étudié à l'Université de Virginie pour son baccalauréat et sa maîtrise, où lui et Yanjun Qi ont effectué des recherches sur les applications de l'apprentissage automatique, en particulier dans la prédiction de la structure des protéines.
Les domaines d'intérêt sont l'apprentissage profond, la prédiction de structure et la biologie de l'information.
Ce qui précède est le contenu détaillé de. pour plus d'informations, suivez d'autres articles connexes sur le site Web de PHP en chinois!

Outils d'IA chauds

Undresser.AI Undress
Application basée sur l'IA pour créer des photos de nu réalistes

AI Clothes Remover
Outil d'IA en ligne pour supprimer les vêtements des photos.

Undress AI Tool
Images de déshabillage gratuites

Clothoff.io
Dissolvant de vêtements AI

AI Hentai Generator
Générez AI Hentai gratuitement.

Article chaud
Outils chauds

Bloc-notes++7.3.1
Éditeur de code facile à utiliser et gratuit

SublimeText3 version chinoise
Version chinoise, très simple à utiliser

Envoyer Studio 13.0.1
Puissant environnement de développement intégré PHP

Dreamweaver CS6
Outils de développement Web visuel

SublimeText3 version Mac
Logiciel d'édition de code au niveau de Dieu (SublimeText3)



 Le robot DeepMind joue au tennis de table, et son coup droit et son revers glissent dans les airs, battant complètement les débutants humains
Aug 09, 2024 pm 04:01 PM
Le robot DeepMind joue au tennis de table, et son coup droit et son revers glissent dans les airs, battant complètement les débutants humains
Aug 09, 2024 pm 04:01 PM
Mais peut-être qu’il ne pourra pas vaincre le vieil homme dans le parc ? Les Jeux Olympiques de Paris battent leur plein et le tennis de table suscite beaucoup d'intérêt. Dans le même temps, les robots ont également réalisé de nouvelles avancées dans le domaine du tennis de table. DeepMind vient tout juste de proposer le premier agent robot apprenant capable d'atteindre le niveau des joueurs amateurs humains de tennis de table de compétition. Adresse papier : https://arxiv.org/pdf/2408.03906 Quelle est la capacité du robot DeepMind à jouer au tennis de table ? Probablement à égalité avec les joueurs amateurs humains : tant en coup droit qu'en revers : l'adversaire utilise une variété de styles de jeu, et le robot peut également résister : recevoir des services avec des tours différents : Cependant, l'intensité du jeu ne semble pas aussi intense que le vieil homme dans le parc. Pour les robots, le tennis de table
 Comprenez la tokenisation en un seul article !
Apr 12, 2024 pm 02:31 PM
Comprenez la tokenisation en un seul article !
Apr 12, 2024 pm 02:31 PM
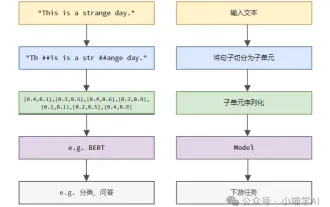
Les modèles de langage raisonnent sur le texte, qui se présente généralement sous la forme de chaînes, mais l'entrée du modèle ne peut être que des nombres, le texte doit donc être converti sous forme numérique. La tokenisation est une tâche fondamentale du traitement du langage naturel. Elle peut diviser une séquence de texte continue (telle que des phrases, des paragraphes, etc.) en une séquence de caractères (telle que des mots, des phrases, des caractères, des signes de ponctuation, etc.) en fonction de besoins spécifiques. Les unités qu'il contient sont appelées un jeton ou un mot. Selon le processus spécifique illustré dans la figure ci-dessous, les phrases de texte sont d'abord divisées en unités, puis les éléments individuels sont numérisés (mappés en vecteurs), puis ces vecteurs sont entrés dans le modèle pour le codage, et enfin sortis vers des tâches en aval pour obtenir en outre le résultat final. La segmentation du texte peut être divisée en Toke en fonction de la granularité de la segmentation du texte.
 Afin de fournir un nouveau système de référence et d'évaluation de questions-réponses scientifiques et complexes pour les grands modèles, l'UNSW, Argonne, l'Université de Chicago et d'autres institutions ont lancé conjointement le cadre SciQAG.
Jul 25, 2024 am 06:42 AM
Afin de fournir un nouveau système de référence et d'évaluation de questions-réponses scientifiques et complexes pour les grands modèles, l'UNSW, Argonne, l'Université de Chicago et d'autres institutions ont lancé conjointement le cadre SciQAG.
Jul 25, 2024 am 06:42 AM
L'ensemble de données ScienceAI Question Answering (QA) joue un rôle essentiel dans la promotion de la recherche sur le traitement du langage naturel (NLP). Des ensembles de données d'assurance qualité de haute qualité peuvent non seulement être utilisés pour affiner les modèles, mais également évaluer efficacement les capacités des grands modèles linguistiques (LLM), en particulier la capacité à comprendre et à raisonner sur les connaissances scientifiques. Bien qu’il existe actuellement de nombreux ensembles de données scientifiques d’assurance qualité couvrant la médecine, la chimie, la biologie et d’autres domaines, ces ensembles de données présentent encore certaines lacunes. Premièrement, le formulaire de données est relativement simple, et la plupart sont des questions à choix multiples. Elles sont faciles à évaluer, mais limitent la plage de sélection des réponses du modèle et ne peuvent pas tester pleinement la capacité du modèle à répondre aux questions scientifiques. En revanche, les questions et réponses ouvertes
 Trois secrets pour déployer de grands modèles dans le cloud
Apr 24, 2024 pm 03:00 PM
Trois secrets pour déployer de grands modèles dans le cloud
Apr 24, 2024 pm 03:00 PM
Compilation|Produit par Xingxuan|51CTO Technology Stack (ID WeChat : blog51cto) Au cours des deux dernières années, j'ai été davantage impliqué dans des projets d'IA générative utilisant de grands modèles de langage (LLM) plutôt que des systèmes traditionnels. Le cloud computing sans serveur commence à me manquer. Leurs applications vont de l’amélioration de l’IA conversationnelle à la fourniture de solutions d’analyse complexes pour diverses industries, ainsi que de nombreuses autres fonctionnalités. De nombreuses entreprises déploient ces modèles sur des plates-formes cloud, car les fournisseurs de cloud public fournissent déjà un écosystème prêt à l'emploi et constituent la voie de moindre résistance. Cependant, cela n’est pas bon marché. Le cloud offre également d'autres avantages tels que l'évolutivité, l'efficacité et des capacités informatiques avancées (GPU disponibles sur demande). Il existe certains aspects peu connus du déploiement de LLM sur les plateformes de cloud public
 Ajustement efficace des paramètres des modèles de langage à grande échelle - Série de réglage fin BitFit/Prefix/Prompt
Oct 07, 2023 pm 12:13 PM
Ajustement efficace des paramètres des modèles de langage à grande échelle - Série de réglage fin BitFit/Prefix/Prompt
Oct 07, 2023 pm 12:13 PM
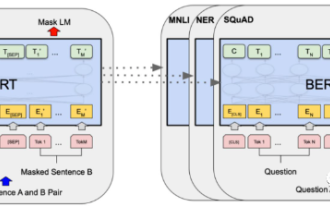
En 2018, Google a publié BERT. Une fois publié, il a vaincu les résultats de pointe (Sota) de 11 tâches PNL d'un seul coup, devenant ainsi une nouvelle étape dans le monde de la PNL. dans la figure ci-dessous, à gauche se trouve le préréglage du modèle BERT, à droite le processus de réglage fin pour des tâches spécifiques. Parmi eux, l'étape de réglage fin est destinée au réglage fin lorsqu'il est ensuite utilisé dans certaines tâches en aval, telles que la classification de texte, le balisage de parties de discours, les systèmes de questions et réponses, etc. BERT peut être affiné sur différents tâches sans ajuster la structure. Grâce à la conception des tâches d'un « modèle de langage pré-entraîné + réglage fin des tâches en aval », il apporte de puissants effets de modèle. Depuis lors, le « modèle linguistique de pré-formation + réglage fin des tâches en aval » est devenu la formation dominante dans le domaine de la PNL.
 AlphaFold 3 est lancé, prédisant de manière exhaustive les interactions et les structures des protéines et de toutes les molécules de la vie, avec une précision bien plus grande que jamais
Jul 16, 2024 am 12:08 AM
AlphaFold 3 est lancé, prédisant de manière exhaustive les interactions et les structures des protéines et de toutes les molécules de la vie, avec une précision bien plus grande que jamais
Jul 16, 2024 am 12:08 AM
Editeur | Radis Skin Depuis la sortie du puissant AlphaFold2 en 2021, les scientifiques utilisent des modèles de prédiction de la structure des protéines pour cartographier diverses structures protéiques dans les cellules, découvrir des médicaments et dresser une « carte cosmique » de chaque interaction protéique connue. Tout à l'heure, Google DeepMind a publié le modèle AlphaFold3, capable d'effectuer des prédictions de structure conjointe pour des complexes comprenant des protéines, des acides nucléiques, de petites molécules, des ions et des résidus modifiés. La précision d’AlphaFold3 a été considérablement améliorée par rapport à de nombreux outils dédiés dans le passé (interaction protéine-ligand, interaction protéine-acide nucléique, prédiction anticorps-antigène). Cela montre qu’au sein d’un cadre unique et unifié d’apprentissage profond, il est possible de réaliser
 Idéalement formé le plus grand ViT de l'histoire ? Google met à niveau le modèle de langage visuel PaLI : prend en charge plus de 100 langues
Apr 12, 2023 am 09:31 AM
Idéalement formé le plus grand ViT de l'histoire ? Google met à niveau le modèle de langage visuel PaLI : prend en charge plus de 100 langues
Apr 12, 2023 am 09:31 AM
Les progrès du traitement du langage naturel ces dernières années proviennent en grande partie de modèles de langage à grande échelle. Chaque nouveau modèle publié pousse la quantité de paramètres et de données d'entraînement vers de nouveaux sommets, et en même temps, les classements de référence existants seront abattus ! Par exemple, en avril de cette année, Google a publié le modèle de langage PaLM (Pathways Language Model) composé de 540 milliards de paramètres, qui a surpassé avec succès les humains dans une série de tests de langage et de raisonnement, en particulier ses excellentes performances dans des scénarios d'apprentissage sur petits échantillons. PaLM est considéré comme la direction de développement du modèle de langage de nouvelle génération. De la même manière, les modèles de langage visuel font des merveilles et les performances peuvent être améliorées en augmentant la taille du modèle. Bien sûr, s'il ne s'agit que d'un modèle de langage visuel multitâche
 RoSA : une nouvelle méthode pour un réglage fin efficace des paramètres de grands modèles
Jan 18, 2024 pm 05:27 PM
RoSA : une nouvelle méthode pour un réglage fin efficace des paramètres de grands modèles
Jan 18, 2024 pm 05:27 PM
À mesure que les modèles de langage évoluent à une échelle sans précédent, un réglage précis des tâches en aval devient prohibitif. Afin de résoudre ce problème, les chercheurs ont commencé à s’intéresser à la méthode PEFT et à l’adopter. L'idée principale de la méthode PEFT est de limiter la portée du réglage fin à un petit ensemble de paramètres afin de réduire les coûts de calcul tout en atteignant des performances de pointe sur les tâches de compréhension du langage naturel. De cette manière, les chercheurs peuvent économiser des ressources informatiques tout en maintenant des performances élevées, ouvrant ainsi la voie à de nouveaux points chauds de recherche dans le domaine du traitement du langage naturel. RoSA est une nouvelle technique PEFT qui, grâce à des expériences sur un ensemble de références, s'est avérée surpasser les précédentes méthodes adaptatives de bas rang (LoRA) et de réglage fin clairsemé pur utilisant le même budget de paramètres. Cet article approfondira
