
 Périphériques technologiques
IA
Stanford et Microsoft se sont associés pour utiliser des modèles de diffusion pour générer des structures protéiques, ce qui est open source
Périphériques technologiques
IA
Stanford et Microsoft se sont associés pour utiliser des modèles de diffusion pour générer des structures protéiques, ce qui est open source
Stanford et Microsoft se sont associés pour utiliser des modèles de diffusion pour générer des structures protéiques, ce qui est open source

Les protéines sont vitales à la vie et jouent un rôle dans presque tous les processus biologiques. D’une part, ils peuvent transmettre des signaux entre neurones, identifier des envahisseurs microscopiques et activer des réponses immunitaires. D’autre part, les protéines ont été largement étudiées comme médiateurs thérapeutiques dans le cadre du traitement de maladies. Ainsi, en générant de nouvelles structures protéiques physiquement pliables, la porte est ouverte à de nouvelles façons d’exploiter les voies cellulaires pour traiter les maladies.
Dans cet article, des chercheurs de l'Université de Stanford, de Microsoft Research et d'autres institutions, inspirés par le processus de repliement des protéines in vivo, ont introduit un modèle de diffusion de repliement (folding diffusion, FoldingDiff), qui reflète les processus naturels de repliement des protéines pour concevoir des protéines. structures de base.

- Adresse papier : https://arxiv.org/pdf/2209.15611.pdf
- Adresse code : https://github.com/microsoft/foldingd si et si
Plus précisément, ils décrivent la structure du squelette protéique comme une série continue d'angles pour capturer les orientations relatives des résidus d'acides aminés constitutifs, et le déplacement inhérent et l'invariance en rotation de cette représentation sont extrêmes. atténue le besoin de réseaux équivariants complexes.
Cette étude a formé un modèle probabiliste de diffusion débruité basé sur le squelette du transformateur et a démontré que notre modèle peut générer de manière inconditionnelle des structures protéiques très réalistes avec une complexité et des modèles structurels similaires aux protéines natives.

Certains internautes ont dit : Je me demande si ce modèle apportera une certaine concurrence à AlphaFold.

Méthodes et résultats
Nous pouvons comprendre les protéines comme des chaînes de résidus d'acides aminés de longueur variable. Il existe 20 acides aminés typiques, partageant le même squelette N-C_α-C à trois atomes, mais avec des côtés différents. les chaînes sont attachées à l’atome C_α (généralement noté R, voir Figure 1).
Ces résidus s'assemblent pour former des chaînes polymères qui se replient en structures 3D dont la forme détermine en grande partie la fonction de la protéine. Ces structures repliées peuvent être décrites à l'aide de quatre niveaux :
- structure primaire, qui capture simplement la séquence linéaire des acides aminés ;
- structure secondaire, qui décrit l'arrangement local des acides aminés ; , Décrit la disposition spatiale complète de tous les résidus ;
- structure quaternaire, qui décrit comment plusieurs chaînes d'acides aminés différentes se réunissent pour former un complexe plus grand.
- Cette étude propose un cadre simplifié de squelette protéique qui suit le processus biologique de repliement des protéines tout en éliminant le besoin de réseaux équivariants complexes. Plutôt que de considérer un squelette protéique, long de N acides aminés, comme une coordonnée tridimensionnelle, ils l’ont vu comme une séquence de six angles internes consécutifs. Autrement dit, étant donné la position du résidu actuel, le vecteur des six angles intérieurs décrit les positions relatives de tous les atomes du squelette dans le résidu suivant. Ces angles intérieurs peuvent être facilement calculés à l’aide de fonctions trigonométriques, en ajoutant de manière itérative des atomes au squelette protéique, puis en les reconvertissant en coordonnées cartésiennes 3D.
L'image ci-dessous montre les résultats d'une expérience. Le diagramme de Ramachandran de la structure naturelle (figure a) contient trois régions correspondant à l'hélice α LH, à l'hélice α RH et au feuillet β. Les trois régions sont entièrement reproduites dans la structure générée ici (Fig. 3b). En d’autres termes, FoldingDiff est capable de générer des éléments de structure secondaires au sein du squelette protéique. De plus, des expériences montrent que le modèle FoldingDiff apprend correctement que les hélices α RH sont plus courantes que les hélices α LH. Les travaux antérieurs utilisant des réseaux équivariants n'ont pas permis de distinguer ces deux types de spirales.

L'image ci-dessous montre l'histogramme bidimensionnel de la structure secondaire dans la chaîne principale de test (4a) et la chaîne principale générée (4b). Les résultats montrent que la structure générée reflète la vraie structure. de la protéine, avec plusieurs hélices alpha, plusieurs feuilles bêta et un mélange des deux.

La figure ci-dessous montre que 111 des 780 structures générées (14,2 %) sont concevables avec un score scTM ≥0,5 (Fig. 5a), ce qui est supérieur à celui rapporté par Trippe et al. 11,8%. Nous voyons également que les chaînes principales générées sont plus similaires aux exemples de formation et ont tendance à avoir une meilleure conception (5b).

Pour plus d'informations, veuillez lire l'article original.
Ce qui précède est le contenu détaillé de. pour plus d'informations, suivez d'autres articles connexes sur le site Web de PHP en chinois!

Outils d'IA chauds

Undresser.AI Undress
Application basée sur l'IA pour créer des photos de nu réalistes

AI Clothes Remover
Outil d'IA en ligne pour supprimer les vêtements des photos.

Undress AI Tool
Images de déshabillage gratuites

Clothoff.io
Dissolvant de vêtements AI

AI Hentai Generator
Générez AI Hentai gratuitement.

Article chaud
Outils chauds

Bloc-notes++7.3.1
Éditeur de code facile à utiliser et gratuit

SublimeText3 version chinoise
Version chinoise, très simple à utiliser

Envoyer Studio 13.0.1
Puissant environnement de développement intégré PHP

Dreamweaver CS6
Outils de développement Web visuel

SublimeText3 version Mac
Logiciel d'édition de code au niveau de Dieu (SublimeText3)



 Le modèle MoE open source le plus puissant au monde est ici, avec des capacités chinoises comparables à celles du GPT-4, et le prix ne représente que près d'un pour cent de celui du GPT-4-Turbo.
May 07, 2024 pm 04:13 PM
Le modèle MoE open source le plus puissant au monde est ici, avec des capacités chinoises comparables à celles du GPT-4, et le prix ne représente que près d'un pour cent de celui du GPT-4-Turbo.
May 07, 2024 pm 04:13 PM
Imaginez un modèle d'intelligence artificielle qui non seulement a la capacité de surpasser l'informatique traditionnelle, mais qui permet également d'obtenir des performances plus efficaces à moindre coût. Ce n'est pas de la science-fiction, DeepSeek-V2[1], le modèle MoE open source le plus puissant au monde est ici. DeepSeek-V2 est un puissant mélange de modèle de langage d'experts (MoE) présentant les caractéristiques d'une formation économique et d'une inférence efficace. Il est constitué de 236B paramètres, dont 21B servent à activer chaque marqueur. Par rapport à DeepSeek67B, DeepSeek-V2 offre des performances plus élevées, tout en économisant 42,5 % des coûts de formation, en réduisant le cache KV de 93,3 % et en augmentant le débit de génération maximal à 5,76 fois. DeepSeek est une entreprise explorant l'intelligence artificielle générale
 L'IA bouleverse la recherche mathématique ! Le lauréat de la médaille Fields et mathématicien sino-américain a dirigé 11 articles les mieux classés | Aimé par Terence Tao
Apr 09, 2024 am 11:52 AM
L'IA bouleverse la recherche mathématique ! Le lauréat de la médaille Fields et mathématicien sino-américain a dirigé 11 articles les mieux classés | Aimé par Terence Tao
Apr 09, 2024 am 11:52 AM
L’IA change effectivement les mathématiques. Récemment, Tao Zhexuan, qui a prêté une attention particulière à cette question, a transmis le dernier numéro du « Bulletin de l'American Mathematical Society » (Bulletin de l'American Mathematical Society). En se concentrant sur le thème « Les machines changeront-elles les mathématiques ? », de nombreux mathématiciens ont exprimé leurs opinions. L'ensemble du processus a été plein d'étincelles, intense et passionnant. L'auteur dispose d'une équipe solide, comprenant Akshay Venkatesh, lauréat de la médaille Fields, le mathématicien chinois Zheng Lejun, l'informaticien de l'Université de New York Ernest Davis et de nombreux autres universitaires bien connus du secteur. Le monde de l’IA a radicalement changé. Vous savez, bon nombre de ces articles ont été soumis il y a un an.
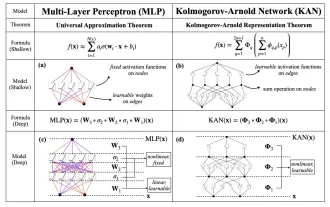 KAN, qui remplace MLP, a été étendu à la convolution par des projets open source
Jun 01, 2024 pm 10:03 PM
KAN, qui remplace MLP, a été étendu à la convolution par des projets open source
Jun 01, 2024 pm 10:03 PM
Plus tôt ce mois-ci, des chercheurs du MIT et d'autres institutions ont proposé une alternative très prometteuse au MLP – KAN. KAN surpasse MLP en termes de précision et d’interprétabilité. Et il peut surpasser le MLP fonctionnant avec un plus grand nombre de paramètres avec un très petit nombre de paramètres. Par exemple, les auteurs ont déclaré avoir utilisé KAN pour reproduire les résultats de DeepMind avec un réseau plus petit et un degré d'automatisation plus élevé. Plus précisément, le MLP de DeepMind compte environ 300 000 paramètres, tandis que le KAN n'en compte qu'environ 200. KAN a une base mathématique solide comme MLP est basé sur le théorème d'approximation universelle, tandis que KAN est basé sur le théorème de représentation de Kolmogorov-Arnold. Comme le montre la figure ci-dessous, KAN a
 Bonjour, Atlas électrique ! Le robot Boston Dynamics revient à la vie, des mouvements étranges à 180 degrés effraient Musk
Apr 18, 2024 pm 07:58 PM
Bonjour, Atlas électrique ! Le robot Boston Dynamics revient à la vie, des mouvements étranges à 180 degrés effraient Musk
Apr 18, 2024 pm 07:58 PM
Boston Dynamics Atlas entre officiellement dans l’ère des robots électriques ! Hier, l'Atlas hydraulique s'est retiré "en larmes" de la scène de l'histoire. Aujourd'hui, Boston Dynamics a annoncé que l'Atlas électrique était au travail. Il semble que dans le domaine des robots humanoïdes commerciaux, Boston Dynamics soit déterminé à concurrencer Tesla. Après la sortie de la nouvelle vidéo, elle a déjà été visionnée par plus d’un million de personnes en seulement dix heures. Les personnes âgées partent et de nouveaux rôles apparaissent. C'est une nécessité historique. Il ne fait aucun doute que cette année est l’année explosive des robots humanoïdes. Les internautes ont commenté : Les progrès des robots ont fait ressembler la cérémonie d'ouverture de cette année à des êtres humains, et le degré de liberté est bien plus grand que celui des humains. Mais n'est-ce vraiment pas un film d'horreur ? Au début de la vidéo, Atlas est allongé calmement sur le sol, apparemment sur le dos. Ce qui suit est à couper le souffle
 Google est ravi : les performances de JAX surpassent Pytorch et TensorFlow ! Cela pourrait devenir le choix le plus rapide pour la formation à l'inférence GPU
Apr 01, 2024 pm 07:46 PM
Google est ravi : les performances de JAX surpassent Pytorch et TensorFlow ! Cela pourrait devenir le choix le plus rapide pour la formation à l'inférence GPU
Apr 01, 2024 pm 07:46 PM
Les performances de JAX, promu par Google, ont dépassé celles de Pytorch et TensorFlow lors de récents tests de référence, se classant au premier rang sur 7 indicateurs. Et le test n’a pas été fait sur le TPU présentant les meilleures performances JAX. Bien que parmi les développeurs, Pytorch soit toujours plus populaire que Tensorflow. Mais à l’avenir, des modèles plus volumineux seront peut-être formés et exécutés sur la base de la plate-forme JAX. Modèles Récemment, l'équipe Keras a comparé trois backends (TensorFlow, JAX, PyTorch) avec l'implémentation native de PyTorch et Keras2 avec TensorFlow. Premièrement, ils sélectionnent un ensemble de
 Recommandé : Excellent projet de détection et de reconnaissance des visages open source JS
Apr 03, 2024 am 11:55 AM
Recommandé : Excellent projet de détection et de reconnaissance des visages open source JS
Apr 03, 2024 am 11:55 AM
La technologie de détection et de reconnaissance des visages est déjà une technologie relativement mature et largement utilisée. Actuellement, le langage d'application Internet le plus utilisé est JS. La mise en œuvre de la détection et de la reconnaissance faciale sur le front-end Web présente des avantages et des inconvénients par rapport à la reconnaissance faciale back-end. Les avantages incluent la réduction de l'interaction réseau et de la reconnaissance en temps réel, ce qui réduit considérablement le temps d'attente des utilisateurs et améliore l'expérience utilisateur. Les inconvénients sont les suivants : il est limité par la taille du modèle et la précision est également limitée ; Comment utiliser js pour implémenter la détection de visage sur le web ? Afin de mettre en œuvre la reconnaissance faciale sur le Web, vous devez être familier avec les langages et technologies de programmation associés, tels que JavaScript, HTML, CSS, WebRTC, etc. Dans le même temps, vous devez également maîtriser les technologies pertinentes de vision par ordinateur et d’intelligence artificielle. Il convient de noter qu'en raison de la conception du côté Web
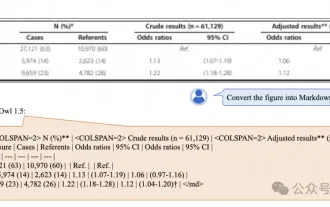 Le document multimodal Alibaba 7B comprenant le grand modèle remporte le nouveau SOTA
Apr 02, 2024 am 11:31 AM
Le document multimodal Alibaba 7B comprenant le grand modèle remporte le nouveau SOTA
Apr 02, 2024 am 11:31 AM
Nouveau SOTA pour des capacités de compréhension de documents multimodaux ! L'équipe Alibaba mPLUG a publié le dernier travail open source mPLUG-DocOwl1.5, qui propose une série de solutions pour relever les quatre défis majeurs que sont la reconnaissance de texte d'image haute résolution, la compréhension générale de la structure des documents, le suivi des instructions et l'introduction de connaissances externes. Sans plus tarder, examinons d’abord les effets. Reconnaissance et conversion en un clic de graphiques aux structures complexes au format Markdown : Des graphiques de différents styles sont disponibles : Une reconnaissance et un positionnement de texte plus détaillés peuvent également être facilement traités : Des explications détaillées sur la compréhension du document peuvent également être données : Vous savez, « Compréhension du document " est actuellement un scénario important pour la mise en œuvre de grands modèles linguistiques. Il existe de nombreux produits sur le marché pour aider à la lecture de documents. Certains d'entre eux utilisent principalement des systèmes OCR pour la reconnaissance de texte et coopèrent avec LLM pour le traitement de texte.
 Les robots Tesla travaillent dans les usines, Musk : Le degré de liberté des mains atteindra 22 cette année !
May 06, 2024 pm 04:13 PM
Les robots Tesla travaillent dans les usines, Musk : Le degré de liberté des mains atteindra 22 cette année !
May 06, 2024 pm 04:13 PM
La dernière vidéo du robot Optimus de Tesla est sortie, et il peut déjà fonctionner en usine. À vitesse normale, il trie les batteries (les batteries 4680 de Tesla) comme ceci : Le responsable a également publié à quoi cela ressemble à une vitesse 20 fois supérieure - sur un petit "poste de travail", en sélectionnant et en sélectionnant et en sélectionnant : Cette fois, il est publié L'un des points forts de la vidéo est qu'Optimus réalise ce travail en usine, de manière totalement autonome, sans intervention humaine tout au long du processus. Et du point de vue d'Optimus, il peut également récupérer et placer la batterie tordue, en se concentrant sur la correction automatique des erreurs : concernant la main d'Optimus, le scientifique de NVIDIA Jim Fan a donné une évaluation élevée : la main d'Optimus est l'un des robots à cinq doigts du monde. le plus adroit. Ses mains ne sont pas seulement tactiles
