
 Périphériques technologiques
IA
L'équipe Tsinghua propose un cadre de pré-formation sur les graphes guidés par les connaissances : une méthode pour améliorer l'apprentissage des représentations moléculaires
Périphériques technologiques
IA
L'équipe Tsinghua propose un cadre de pré-formation sur les graphes guidés par les connaissances : une méthode pour améliorer l'apprentissage des représentations moléculaires
L'équipe Tsinghua propose un cadre de pré-formation sur les graphes guidés par les connaissances : une méthode pour améliorer l'apprentissage des représentations moléculaires


Pour faciliter la prédiction des propriétés moléculaires, il est très important d'apprendre une représentation efficace des caractéristiques moléculaires dans le domaine de la découverte de médicaments. Récemment, les gens ont surmonté le défi de la rareté des données en pré-entraînant les réseaux de neurones graphiques (GNN) à l'aide de techniques d'apprentissage auto-supervisées. Cependant, les méthodes actuelles basées sur l'apprentissage auto-supervisé présentent deux problèmes principaux : le manque de stratégies claires d'apprentissage auto-supervisé et les capacités limitées du GNN
Récemment, des équipes de recherche de l'Université Tsinghua, de l'Université de West Lake et du laboratoire Zhijiang ont proposé des connaissances. conseils Pré-formation guidée par les connaissances de Graph Transformer (KPGT), un cadre d'apprentissage auto-supervisé qui fournit des prédictions améliorées, généralisables et robustes des propriétés moléculaires grâce à un apprentissage de représentation moléculaire considérablement amélioré. Le framework KPGT intègre un transformateur de graphes conçu spécifiquement pour les graphes moléculaires et une stratégie de pré-formation guidée par les connaissances pour capturer pleinement les connaissances structurelles et sémantiques des molécules.
Grâce à des tests informatiques approfondis sur 63 ensembles de données, KPGT a démontré des performances supérieures dans la prédiction des propriétés moléculaires dans divers domaines. En outre, l'applicabilité pratique du KPGT dans la découverte de médicaments a été vérifiée en identifiant des inhibiteurs potentiels de deux cibles antitumorales. Dans l’ensemble, KPGT peut constituer un outil puissant et utile pour faire progresser le processus de découverte de médicaments assisté par l’IA.
La recherche s'intitulait « Un cadre de pré-formation guidé par les connaissances pour améliorer l'apprentissage des représentations moléculaires » et a été publiée dans « Nature Communications » le 21 novembre 2023.

La détermination expérimentale des propriétés moléculaires nécessite beaucoup de temps et de ressources, et l'identification de molécules possédant les propriétés souhaitées est l'un des défis les plus importants de la découverte de médicaments. Ces dernières années, les méthodes basées sur l’intelligence artificielle ont joué un rôle de plus en plus important dans la prédiction des propriétés moléculaires. L'un des principaux défis des méthodes basées sur l'intelligence artificielle pour prédire les propriétés moléculaires est la caractérisation des molécules
Ces dernières années, les méthodes basées sur l'apprentissage profond sont apparues comme des outils potentiellement utiles pour prédire les propriétés moléculaires, principalement en raison de leur capacité à extraire automatiquement à partir de données d’entrée simples Capacité supérieure à caractériser efficacement. Notamment, diverses architectures de réseaux de neurones, notamment les réseaux de neurones récurrents (RNN), les réseaux de neurones convolutifs (CNN) et les réseaux de neurones graphiques (GNN), sont capables de modéliser des données moléculaires dans divers formats, allant des entrées moléculaires simplifiées au système d'entrée de ligne ( SMILES) aux images moléculaires et aux diagrammes moléculaires. Cependant, la disponibilité limitée des molécules marqueurs et l’immensité de l’espace chimique limitent leurs performances prédictives, en particulier lorsqu’il s’agit d’échantillons de données non distribués.
Grâce aux réalisations remarquables des méthodes d'apprentissage auto-supervisées dans les domaines du traitement du langage naturel et de la vision par ordinateur, ces techniques ont été appliquées pour pré-entraîner les GNN et améliorer l'apprentissage des représentations de molécules, obtenant ainsi des résultats substantiels dans les tâches de prédiction des propriétés moléculaires en aval. . Progrès
Les chercheurs émettent l'hypothèse que l'introduction de connaissances supplémentaires décrivant quantitativement les caractéristiques moléculaires dans un cadre d'apprentissage auto-supervisé peut efficacement relever ces défis. Les molécules possèdent de nombreuses caractéristiques quantitatives, telles que des descripteurs moléculaires et des empreintes digitales, qui peuvent être facilement obtenues avec les outils informatiques actuellement établis. L'intégration de ces connaissances supplémentaires peut introduire de riches informations sémantiques moléculaires dans l'apprentissage auto-supervisé, améliorant ainsi considérablement l'acquisition de représentations moléculaires sémantiquement riches.
De manière générale, les méthodes d'apprentissage auto-supervisées existantes s'appuient sur GNN comme modèle de base. Cependant, GNN a une capacité de modèle limitée. De plus, les GNN peuvent avoir des difficultés à capturer les interactions à longue portée entre les atomes. Et les modèles basés sur Transformer sont devenus un modèle révolutionnaire. Il se caractérise par un nombre croissant de paramètres et la capacité de capturer des interactions à longue portée, offrant une approche prometteuse pour modéliser de manière exhaustive les caractéristiques structurelles des molécules
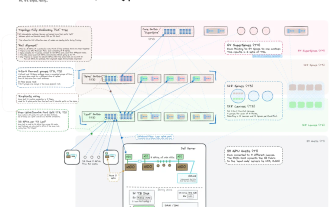
Cadre d'apprentissage auto-supervisé KPGT
Dans cette étude, les chercheurs ont introduit Un cadre d'apprentissage auto-supervisé appelé KPGT est développé pour améliorer l'apprentissage de la représentation moléculaire et promouvoir ainsi les tâches de prédiction des propriétés moléculaires en aval. Le cadre KPGT se compose de deux composants principaux : un modèle de base appelé Line Graph Transformer (LiGhT) et une politique de pré-formation guidée par les connaissances. Le framework KPGT combine le modèle LiGhT haute capacité, spécialement conçu pour modéliser avec précision les structures de graphes moléculaires, et utilise une stratégie de pré-formation guidée par les connaissances pour capturer la structure moléculaire et les connaissances sémantiques.
L'équipe de recherche a utilisé environ 2 millions de dollars du L'ensemble de données ChEMBL29 Molécule, LiGhT a été pré-entraîné via une stratégie de pré-entraînement guidée par les connaissances
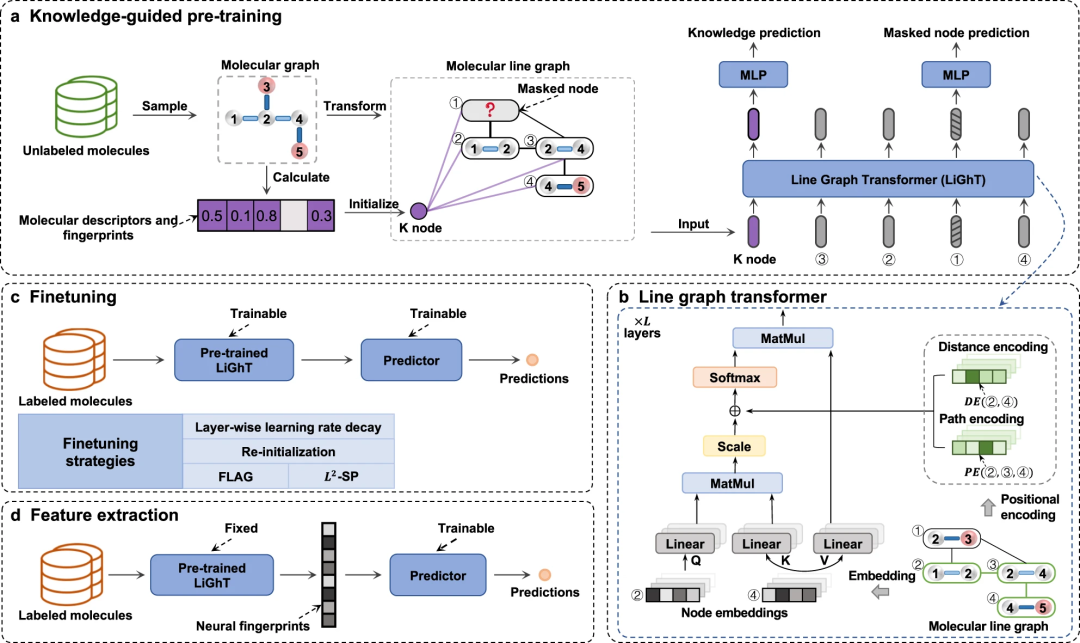
Contenu réécrit : Diagramme : Présentation de KPGT. (Source : article)
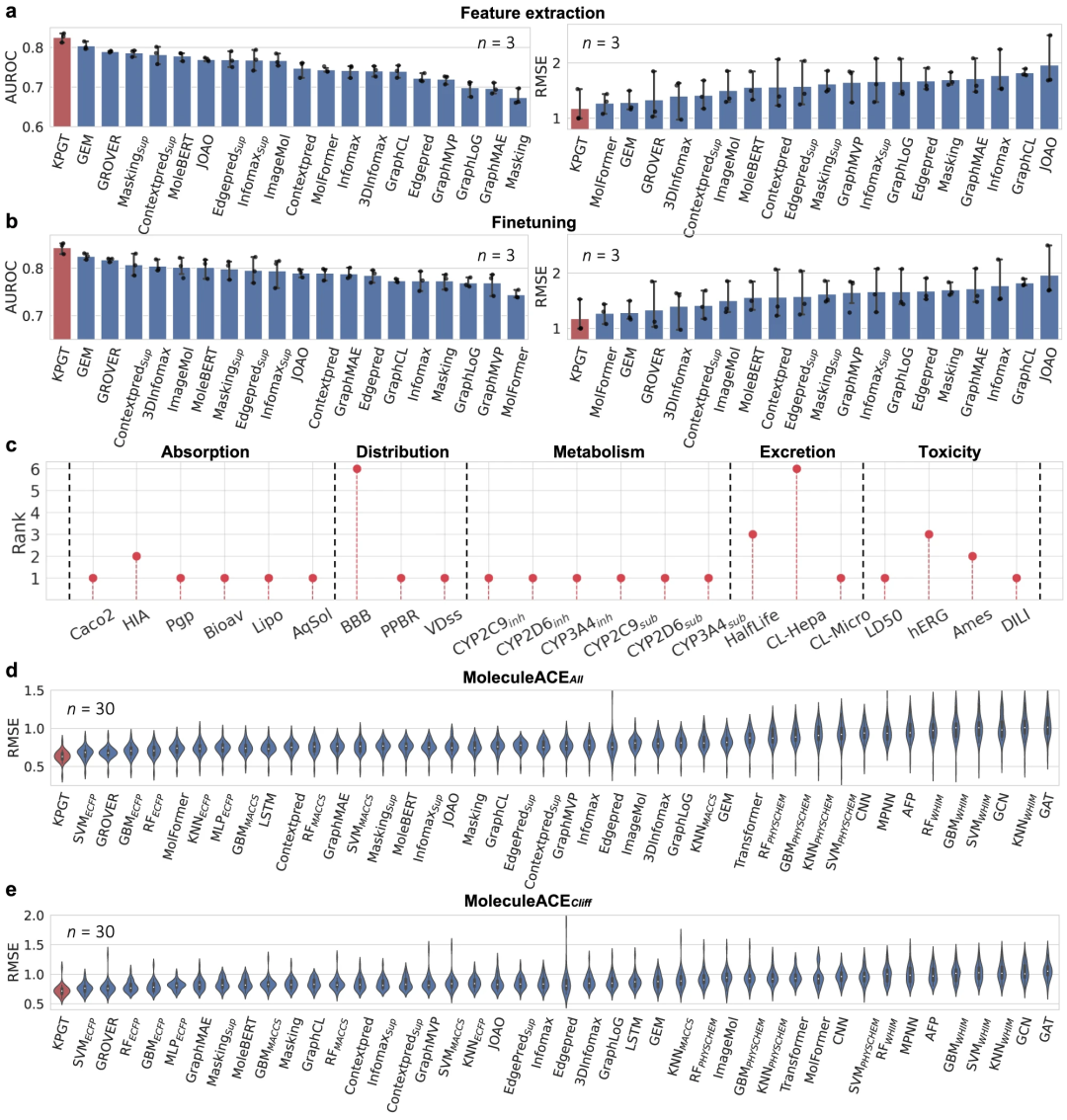
KPGT surpasse les méthodes de base en matière de prédiction des propriétés moléculaires. Par rapport à plusieurs méthodes de base, KPGT réalise des améliorations significatives sur 63 ensembles de données.
De plus, l'application pratique du KPGT a été démontrée en utilisant avec succès le KPGT pour identifier des inhibiteurs potentiels de deux cibles antitumorales, la kinase progénitrice hématopoïétique 1 (HPK1) et le récepteur du facteur de croissance des fibroblastes sexuels (FGFR1).
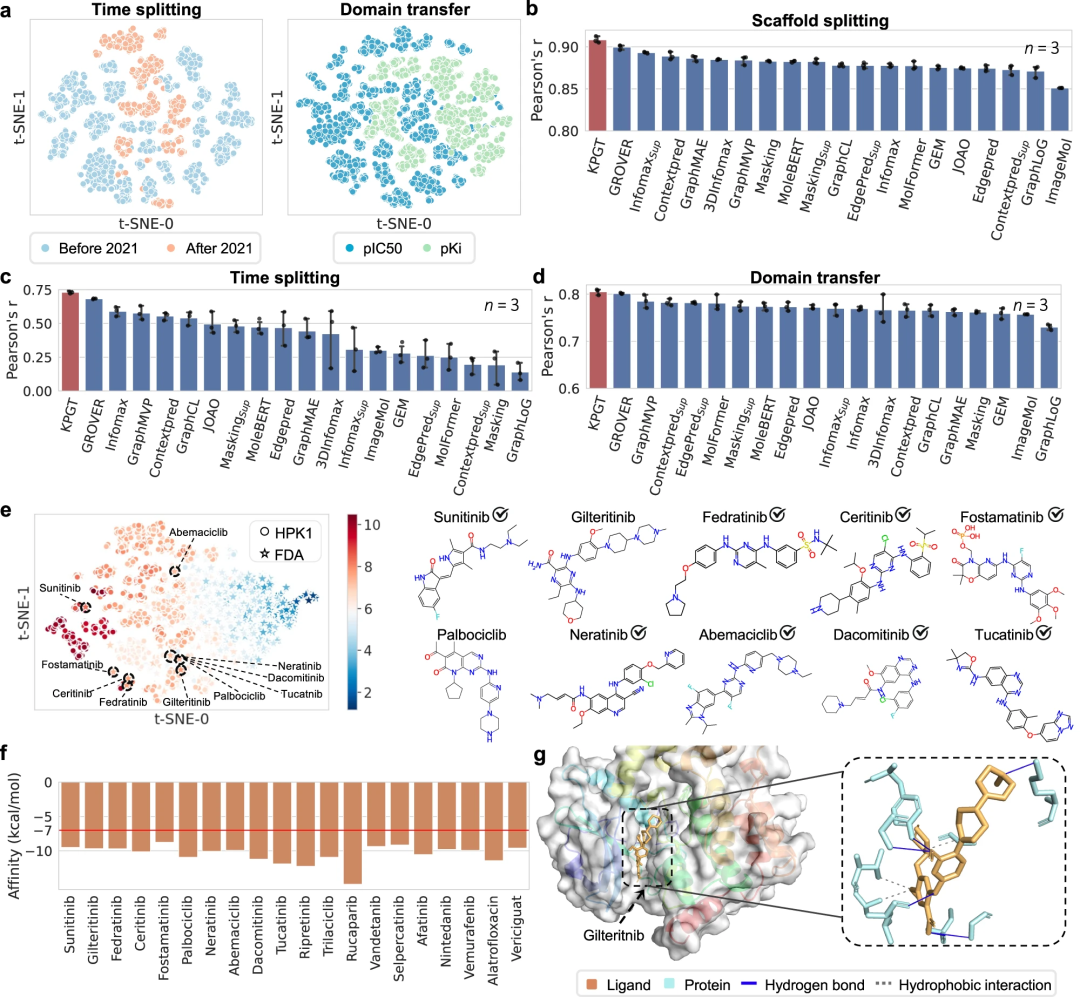
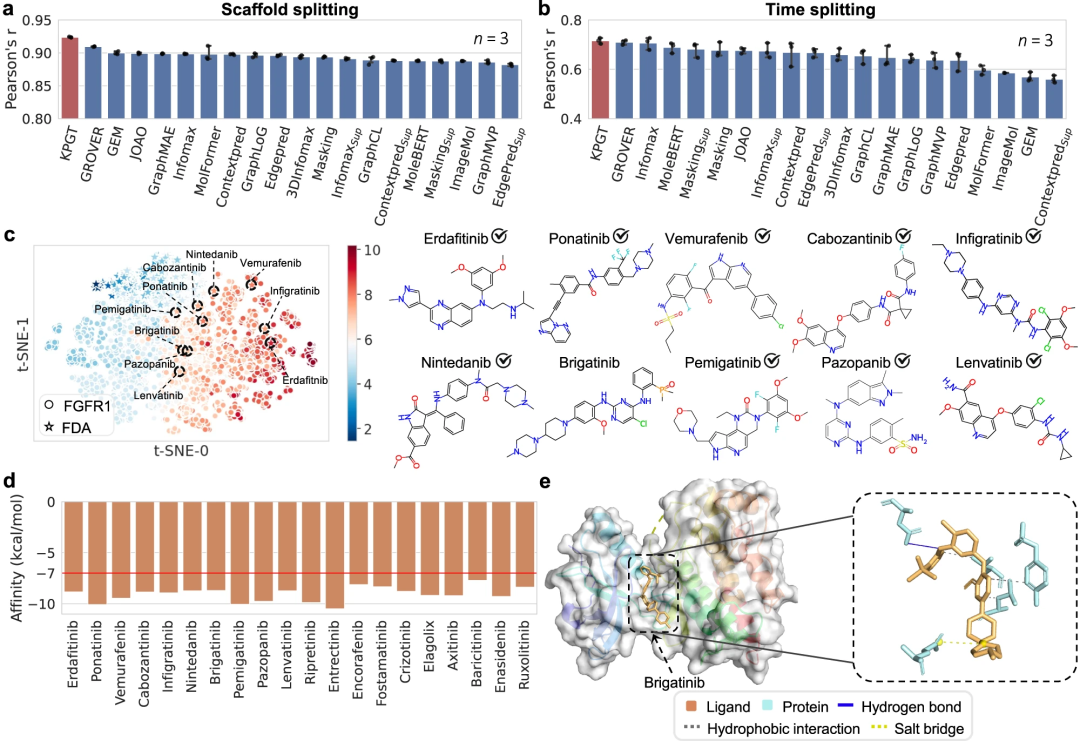
Limites de la recherche
Malgré les avantages du KPGT dans la prédiction efficace des propriétés moléculaires, il existe encore certaines limites.
Tout d'abord, l'intégration de connaissances supplémentaires est la caractéristique la plus significative de la méthode proposée. En plus des 200 descripteurs moléculaires et des 512 RDKFP utilisés dans KPGT, il est possible d'incorporer divers autres types de connaissances complémentaires. De plus, des recherches ultérieures pourraient intégrer des conformations moléculaires tridimensionnelles (3D) dans le processus de pré-entraînement, permettant au modèle de capturer des informations 3D importantes sur la molécule et d'améliorer potentiellement les capacités d'apprentissage des représentations. Bien que KPGT utilise actuellement un modèle de base avec environ 100 millions de paramètres et un pré-entraînement sur 2 millions de molécules, l'exploration d'un pré-entraînement à plus grande échelle pourrait offrir des avantages plus substantiels pour l'apprentissage des représentations moléculaires.
Dans l'ensemble, KPGT fournit un puissant cadre d'apprentissage auto-supervisé pour un apprentissage efficace des représentations moléculaires, faisant ainsi progresser le domaine de la découverte de médicaments assistée par l'intelligence artificielle.
Lien papier : https://www.nature.com/articles/s41467-023-43214-1
Ce qui précède est le contenu détaillé de. pour plus d'informations, suivez d'autres articles connexes sur le site Web de PHP en chinois!

Outils d'IA chauds

Undresser.AI Undress
Application basée sur l'IA pour créer des photos de nu réalistes

AI Clothes Remover
Outil d'IA en ligne pour supprimer les vêtements des photos.

Undress AI Tool
Images de déshabillage gratuites

Clothoff.io
Dissolvant de vêtements AI

AI Hentai Generator
Générez AI Hentai gratuitement.

Article chaud
Outils chauds

Bloc-notes++7.3.1
Éditeur de code facile à utiliser et gratuit

SublimeText3 version chinoise
Version chinoise, très simple à utiliser

Envoyer Studio 13.0.1
Puissant environnement de développement intégré PHP

Dreamweaver CS6
Outils de développement Web visuel

SublimeText3 version Mac
Logiciel d'édition de code au niveau de Dieu (SublimeText3)



 Un didacticiel sur le modèle de diffusion qui vaut votre temps, de l'Université Purdue
Apr 07, 2024 am 09:01 AM
Un didacticiel sur le modèle de diffusion qui vaut votre temps, de l'Université Purdue
Apr 07, 2024 am 09:01 AM
La diffusion permet non seulement de mieux imiter, mais aussi de « créer ». Le modèle de diffusion (DiffusionModel) est un modèle de génération d'images. Par rapport aux algorithmes bien connus tels que GAN et VAE dans le domaine de l’IA, le modèle de diffusion adopte une approche différente. Son idée principale est un processus consistant à ajouter d’abord du bruit à l’image, puis à la débruiter progressivement. Comment débruiter et restaurer l’image originale est la partie centrale de l’algorithme. L'algorithme final est capable de générer une image à partir d'une image bruitée aléatoirement. Ces dernières années, la croissance phénoménale de l’IA générative a permis de nombreuses applications passionnantes dans la génération de texte en image, la génération de vidéos, et bien plus encore. Le principe de base de ces outils génératifs est le concept de diffusion, un mécanisme d'échantillonnage spécial qui surmonte les limites des méthodes précédentes.
 Générez du PPT en un seul clic ! Kimi : Que les « travailleurs migrants PPT » deviennent d'abord populaires
Aug 01, 2024 pm 03:28 PM
Générez du PPT en un seul clic ! Kimi : Que les « travailleurs migrants PPT » deviennent d'abord populaires
Aug 01, 2024 pm 03:28 PM
Kimi : En une seule phrase, un PPT est prêt en seulement dix secondes. PPT est tellement ennuyeux ! Pour tenir une réunion, vous devez avoir un PPT ; pour rédiger un rapport hebdomadaire, vous devez avoir un PPT ; pour solliciter des investissements, vous devez présenter un PPT ; même pour accuser quelqu'un de tricherie, vous devez envoyer un PPT ; L'université ressemble plus à une spécialisation PPT. Vous regardez le PPT en classe et faites le PPT après les cours. Peut-être que lorsque Dennis Austin a inventé le PPT il y a 37 ans, il ne s'attendait pas à ce qu'un jour le PPT devienne aussi répandu. Parler de notre dure expérience de création de PPT nous fait monter les larmes aux yeux. "Il m'a fallu trois mois pour réaliser un PPT de plus de 20 pages, et je l'ai révisé des dizaines de fois. J'avais envie de vomir quand j'ai vu le PPT." "À mon apogée, je faisais cinq PPT par jour, et même ma respiration." était PPT." Si vous avez une réunion impromptue, vous devriez le faire
 Tous les prix CVPR 2024 annoncés ! Près de 10 000 personnes ont assisté à la conférence hors ligne et un chercheur chinois de Google a remporté le prix du meilleur article.
Jun 20, 2024 pm 05:43 PM
Tous les prix CVPR 2024 annoncés ! Près de 10 000 personnes ont assisté à la conférence hors ligne et un chercheur chinois de Google a remporté le prix du meilleur article.
Jun 20, 2024 pm 05:43 PM
Tôt le matin du 20 juin, heure de Pékin, CVPR2024, la plus grande conférence internationale sur la vision par ordinateur qui s'est tenue à Seattle, a officiellement annoncé le meilleur article et d'autres récompenses. Cette année, un total de 10 articles ont remporté des prix, dont 2 meilleurs articles et 2 meilleurs articles étudiants. De plus, il y a eu 2 nominations pour les meilleurs articles et 4 nominations pour les meilleurs articles étudiants. La conférence la plus importante dans le domaine de la vision par ordinateur (CV) est la CVPR, qui attire chaque année un grand nombre d'instituts de recherche et d'universités. Selon les statistiques, un total de 11 532 articles ont été soumis cette année, dont 2 719 ont été acceptés, avec un taux d'acceptation de 23,6 %. Selon l'analyse statistique des données CVPR2024 du Georgia Institute of Technology, du point de vue des sujets de recherche, le plus grand nombre d'articles est la synthèse et la génération d'images et de vidéos (Imageandvideosyn
 Du bare metal au grand modèle avec 70 milliards de paramètres, voici un tutoriel et des scripts prêts à l'emploi
Jul 24, 2024 pm 08:13 PM
Du bare metal au grand modèle avec 70 milliards de paramètres, voici un tutoriel et des scripts prêts à l'emploi
Jul 24, 2024 pm 08:13 PM
Nous savons que le LLM est formé sur des clusters informatiques à grande échelle utilisant des données massives. Ce site a présenté de nombreuses méthodes et technologies utilisées pour aider et améliorer le processus de formation LLM. Aujourd'hui, ce que nous souhaitons partager est un article qui approfondit la technologie sous-jacente et présente comment transformer un ensemble de « bare metals » sans même un système d'exploitation en un cluster informatique pour la formation LLM. Cet article provient d'Imbue, une startup d'IA qui s'efforce d'atteindre une intelligence générale en comprenant comment les machines pensent. Bien sûr, transformer un tas de « bare metal » sans système d'exploitation en un cluster informatique pour la formation LLM n'est pas un processus facile, plein d'exploration et d'essais et d'erreurs, mais Imbue a finalement réussi à former un LLM avec 70 milliards de paramètres et dans. le processus s'accumule
 Guide d'installation de PyCharm Community Edition : maîtrisez rapidement toutes les étapes
Jan 27, 2024 am 09:10 AM
Guide d'installation de PyCharm Community Edition : maîtrisez rapidement toutes les étapes
Jan 27, 2024 am 09:10 AM
Démarrage rapide avec PyCharm Community Edition : Tutoriel d'installation détaillé Analyse complète Introduction : PyCharm est un puissant environnement de développement intégré (IDE) Python qui fournit un ensemble complet d'outils pour aider les développeurs à écrire du code Python plus efficacement. Cet article présentera en détail comment installer PyCharm Community Edition et fournira des exemples de code spécifiques pour aider les débutants à démarrer rapidement. Étape 1 : Téléchargez et installez PyCharm Community Edition Pour utiliser PyCharm, vous devez d'abord le télécharger depuis son site officiel
 Cinq logiciels de programmation pour débuter l'apprentissage du langage C
Feb 19, 2024 pm 04:51 PM
Cinq logiciels de programmation pour débuter l'apprentissage du langage C
Feb 19, 2024 pm 04:51 PM
En tant que langage de programmation largement utilisé, le langage C est l'un des langages de base qui doivent être appris pour ceux qui souhaitent se lancer dans la programmation informatique. Cependant, pour les débutants, l’apprentissage d’un nouveau langage de programmation peut s’avérer quelque peu difficile, notamment en raison du manque d’outils d’apprentissage et de matériel pédagogique pertinents. Dans cet article, je présenterai cinq logiciels de programmation pour aider les débutants à démarrer avec le langage C et vous aider à démarrer rapidement. Le premier logiciel de programmation était Code :: Blocks. Code::Blocks est un environnement de développement intégré (IDE) gratuit et open source pour
 A lire absolument pour les débutants en technique : Analyse des niveaux de difficulté du langage C et Python
Mar 22, 2024 am 10:21 AM
A lire absolument pour les débutants en technique : Analyse des niveaux de difficulté du langage C et Python
Mar 22, 2024 am 10:21 AM
Titre : Une lecture incontournable pour les débutants en technique : Analyse des difficultés du langage C et de Python, nécessitant des exemples de code spécifiques. À l'ère numérique d'aujourd'hui, la technologie de programmation est devenue une capacité de plus en plus importante. Que vous souhaitiez travailler dans des domaines tels que le développement de logiciels, l'analyse de données, l'intelligence artificielle ou simplement apprendre la programmation par intérêt, choisir un langage de programmation adapté est la première étape. Parmi les nombreux langages de programmation, le langage C et Python sont deux langages de programmation largement utilisés, chacun ayant ses propres caractéristiques. Cet article analysera les niveaux de difficulté du langage C et Python
 L'IA utilisée | L'IA a créé un vlog sur la vie d'une fille vivant seule, qui a reçu des dizaines de milliers de likes en 3 jours
Aug 07, 2024 pm 10:53 PM
L'IA utilisée | L'IA a créé un vlog sur la vie d'une fille vivant seule, qui a reçu des dizaines de milliers de likes en 3 jours
Aug 07, 2024 pm 10:53 PM
Rédacteur du Machine Power Report : Yang Wen La vague d’intelligence artificielle représentée par les grands modèles et l’AIGC a discrètement changé notre façon de vivre et de travailler, mais la plupart des gens ne savent toujours pas comment l’utiliser. C'est pourquoi nous avons lancé la rubrique « AI in Use » pour présenter en détail comment utiliser l'IA à travers des cas d'utilisation de l'intelligence artificielle intuitifs, intéressants et concis et stimuler la réflexion de chacun. Nous invitons également les lecteurs à soumettre des cas d'utilisation innovants et pratiques. Lien vidéo : https://mp.weixin.qq.com/s/2hX_i7li3RqdE4u016yGhQ Récemment, le vlog de la vie d'une fille vivant seule est devenu populaire sur Xiaohongshu. Une animation de style illustration, associée à quelques mots de guérison, peut être facilement récupérée en quelques jours seulement.
