
 Périphériques technologiques
IA
L'alchimie de l'IA révolutionne la chimie : des chercheurs du MIT utilisent l'IA générative pour générer de nouvelles réactions chimiques en six secondes
Périphériques technologiques
IA
L'alchimie de l'IA révolutionne la chimie : des chercheurs du MIT utilisent l'IA générative pour générer de nouvelles réactions chimiques en six secondes
L'alchimie de l'IA révolutionne la chimie : des chercheurs du MIT utilisent l'IA générative pour générer de nouvelles réactions chimiques en six secondes


Ce qui doit être réécrit, c'est : Éditeur | Kaixia
La chimie, à partir de l'alchimie ancienne de « l'échange équivalent », a toujours été une discipline qui étudie et contrôle l'interaction entre les substances. En déverrouillant et en exploitant continuellement de nouvelles réactions chimiques, de nombreux nouveaux matériaux ont été développés. Ces nouveaux matériaux apportent non seulement du confort à la vie des gens, mais améliorent également l'efficacité de l'utilisation de l'énergie et favorisent le développement durable. Une réaction chimique de base est constituée de réactifs, d'états de transition (TS) et de produits. Les états de transition sont des structures 3D cruciales en chimie et sont largement utilisés pour comprendre les mécanismes de réaction chimique, estimer les barrières énergétiques de réaction et explorer de vastes réseaux de réactions. Cependant, en raison du temps extrêmement court (ordre femtoseconde) pendant lequel ils existent au cours de la réaction, il est presque impossible d’isoler et de caractériser expérimentalement l’état de transition.
Contenu réécrit : Habituellement, les gens utilisent des méthodes de calcul de chimie quantique pour déterminer l'état de transition entre les réactifs et les produits connus en résolvant à plusieurs reprises l'équation de Schrödinger. Cependant, cette méthode de calcul est très coûteuse et connue pour ses échecs fréquents. Dans le même temps, cette méthode est limitée par l’expérience personnelle, l’intuition et les ressources informatiques, et les réactions chimiques que chaque personne peut explorer sont également limitées. Cette limitation est particulièrement fatale lors de l’étude de réactions complexes inconnues. Cela amènera les chercheurs à ignorer certaines réactions potentielles, jugeant ainsi mal le mécanisme de réaction, affectant ainsi la conception des matériaux catalytiques.
En réponse à ce problème, un groupe de chercheurs du Massachusetts Institute of Technology (MIT) a développé une méthode alternative basée sur l'apprentissage automatique peut découvrir ces structures en quelques secondes. Leur nouveau modèle pourrait aider les chimistes à explorer et à concevoir de nouvelles réactions et catalyseurs pour générer des produits utiles à haute valeur ajoutée, tels que des composés combustibles ou des produits pharmaceutiques. De plus, le modèle est capable de simuler des réactions chimiques naturelles, telles que celles essentielles à l’évolution de la vie sur la Terre primitive.
Heather Kulik, professeur de génie chimique et de chimie au MIT, a souligné que comprendre la structure spécifique de l'état de transition est très important pour concevoir des catalyseurs ou comprendre comment les systèmes naturels effectuent certaines transformations.
Les travaux de recherche connexes sont intitulés "État de transition précis génération avec un « modèle de diffusion de réaction élémentaire équivariant » sensible aux objets a été publié dans la revue internationale de premier plan « Nature Computational Science ».
Le Dr Duan Chenru du MIT est le premier auteur de l'article. Du Yuanqi, doctorant à l'Université Cornell, Jia Haojun, doctorant au MIT, et le professeur Heather Kulik du MIT sont co-auteurs de l'article. . Lien original : [https://rdcu.be/dtGSF]
 Veuillez cliquer sur le lien suivant pour consulter l'article : https://www.nature.com/articles/s43588-023-00563-7
Veuillez cliquer sur le lien suivant pour consulter l'article : https://www.nature.com/articles/s43588-023-00563-7
MIT College News a également rendu compte de cette étude
 Lien de rapport : https://news.mit.edu/2023/computational-model-captures-elusive-transition-states-1215
Lien de rapport : https://news.mit.edu/2023/computational-model-captures-elusive-transition-states-1215
Actuellement, les chimistes peuvent utiliser des méthodes de calcul de chimie quantique basées sur la théorie fonctionnelle de la densité pour calculer les états de transition. Cependant, cette méthode nécessite beaucoup de ressources informatiques, et il faut des heures, voire des jours, pour terminer le calcul d'un état de transition
Afin de résoudre le problème des longs temps de calcul, certains chercheurs ont récemment commencé à essayer d'utiliser l'apprentissage automatique modèles pour découvrir les structures des États de transition. Cependant, presque tous les modèles développés à ce jour nécessitent que les deux réactifs soient modélisés dans leur ensemble, les réactifs conservant une configuration géométrique spécifique l'un par rapport à l'autre. Toute autre configuration possible sera confondue par le modèle d'apprentissage automatique avec une nouvelle réaction.
Le Dr Duan Chenru a déclaré que si les molécules réactives sont mises en rotation, en principe, elles peuvent toujours subir la même réaction chimique avant et après la rotation. Tout comme lorsque nous parlons d’électrolyse de l’eau, nous disons seulement que l’eau est convertie en oxygène et en hydrogène dans des conditions spécifiques, sans décrire les positions géométriques relatives de ces molécules. Mais dans les méthodes traditionnelles d’apprentissage automatique, le modèle traitera les réactions des réactifs et des produits à différentes positions géométriques comme deux réactions différentes. Cela rend la formation en apprentissage automatique plus difficile et la précision diminuera
Le modèle de diffusion est un modèle génératif largement utilisé en traitement d'image. Récemment, des modèles de diffusion ont également été utilisés pour générer des structures moléculaires et protéiques 3D, un amarrage protéine-ligand et une conception de médicaments basée sur la structure. Dans ces applications, les modèles de diffusion utilisent des réseaux de neurones graphiques (GNN) 3D spéciaux du groupe euclidien (SE (3)) pour préserver l'alignement, les symétries de translation et de rotation des molécules. Cependant, les réactions élémentaires sont constituées de réactifs, d'états de transition et de produits et suivent la symétrie SE(3) « sensible aux objets ». En effet, l'interaction entre les trois objets dans la réaction élémentaire ne s'effectue pas dans l'espace euclidien 3D, mais constitue une connexion causale sur la surface d'énergie potentielle électronique de dimension supérieure. Par conséquent, le modèle de diffusion existant basé sur SE(3) GNN peut avoir des problèmes en raison de la destruction de la symétrie
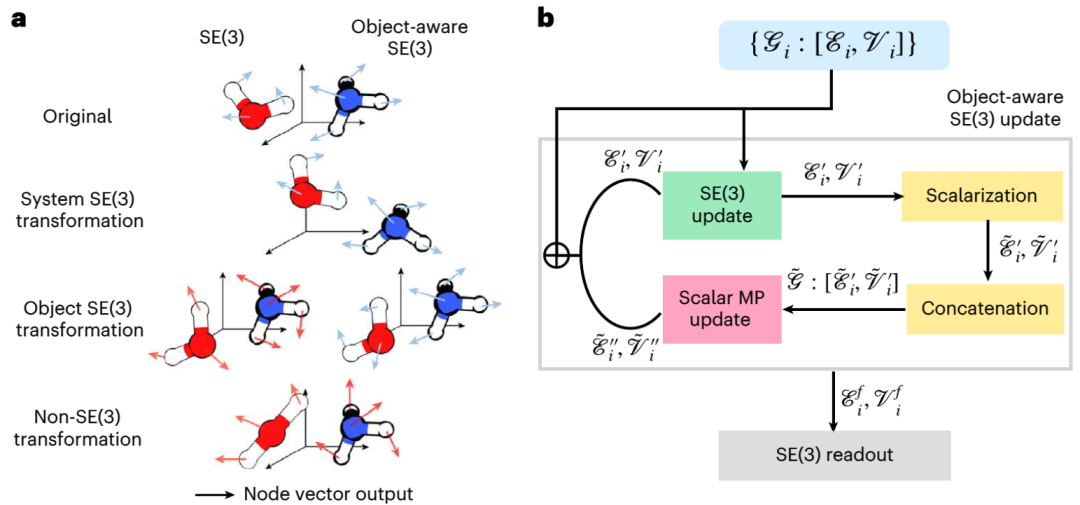
Illustration : "Object Perception" SE(3) équivariance et son équivariance SE(3) basée sur la mise en œuvre de GNN . (Source : Paper)
Solution
L'équipe du MIT a développé une nouvelle méthode appelée « OA-ReactDiff » basée sur les problèmes ci-dessus. L'équipe a ajusté le GNN équivariant SE(3) à une simulation « sensible aux objets », c'est-à-dire en maintenant l'équivariance SE(3) d'un seul objet tout en conservant leurs interactions indépendantes dans l'espace euclidien. Les modèles de diffusion du Dr Duan Chenru en font partie. du domaine de l'intelligence artificielle générative, qui utilise des processus stochastiques pour capturer la transformation entre des distributions simples et complexes. Une fois que le modèle a appris la distribution de base de la façon dont ces trois structures coexistent, nous pouvons lui donner de nouveaux réactifs et produits et il essaiera de générer des structures d'état de transition correspondant à ces réactifs et produits
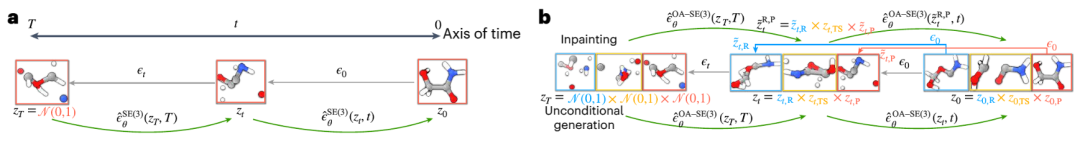 Illustration : Aperçu du modèle de diffusion équivariant (EDM) pour générer des échantillons de systèmes moléculaires. (Source : article)
Illustration : Aperçu du modèle de diffusion équivariant (EDM) pour générer des échantillons de systèmes moléculaires. (Source : article)
Dans l'étude, les chercheurs ont utilisé des méthodes informatiques quantiques pour obtenir les structures des réactifs, les états de transition et les produits de 9 000 réactions chimiques différentes dans l'ensemble d'entraînement. Et a également testé environ 1 000 réactions inédites, nécessitant la génération de 40 structures possibles pour chaque état de transition
Au cours du processus de calcul, un « modèle de recommandation » a été introduit pour prédire le niveau de confiance de quel état de transition est le plus élevé. Sur cette base, combinée en outre avec des estimations d'incertitude, les chercheurs ont effectué des calculs de chimie quantique uniquement sur les 14 % des réactions présentant l'incertitude de modèle la plus élevée, atteignant avec succès une erreur absolue moyenne de 2,6 kcal/mol. Cela permet d'obtenir des résultats dans une erreur d'un ordre de grandeur lors de l'estimation des taux de réaction à 300 °C à l'aide de OA-ReactDiff. Comparée à la structure d'état de transition obtenue par des calculs de chimie quantique, l'erreur quadratique moyenne (RMSD) de la structure générée par OA-ReactDiff est de l'ordre de 0,06 angström (six millièmes de nanomètre), une ampleur d'erreur presque impossible à distinguer. à l'œil nu
Ce qui est encore plus gratifiant, c'est qu'OA-ReactDiff ne prend que 6 secondes pour générer une structure d'état de transition, ce qui est au moins 1000 fois plus rapide que les calculs de chimie quantique. En conséquence, l’algorithme atteint avec succès une précision et une rapidité extrêmement élevées dans le calcul des structures TS et des barrières d’énergie de réaction.
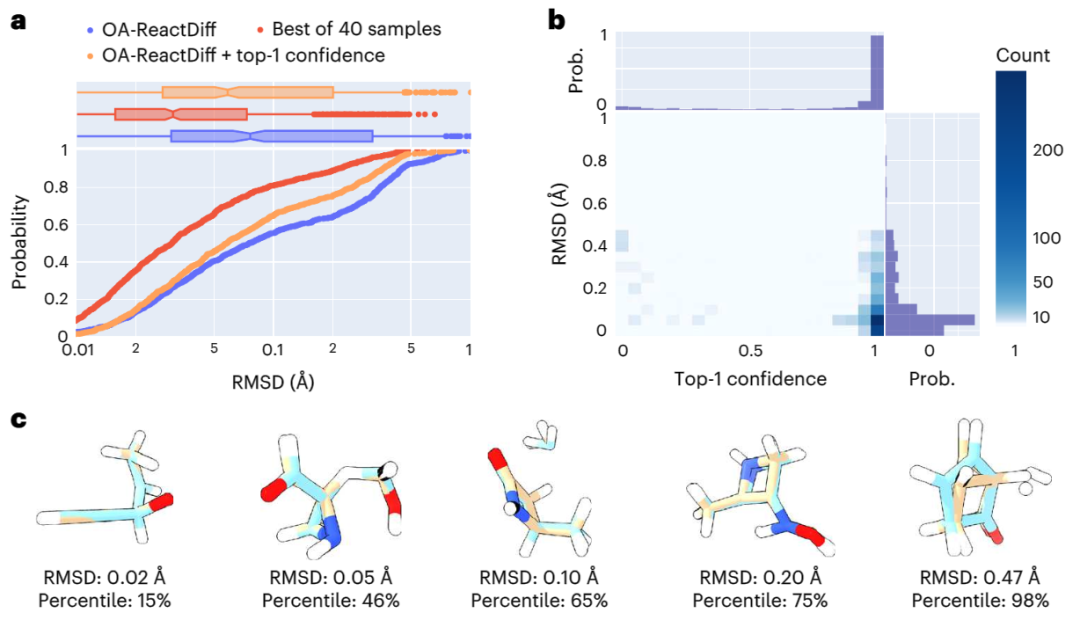 Illustration : Évaluation de la similitude structurelle entre la structure TS générée par OA-ReactDiff et la structure TS réelle. (Source : article)
Illustration : Évaluation de la similitude structurelle entre la structure TS générée par OA-ReactDiff et la structure TS réelle. (Source : article)
Le professeur Kulik a également déploré : « Il était difficile pour nous d'imaginer que des milliers d'états de transition puissent être générés en une seule pensée. »
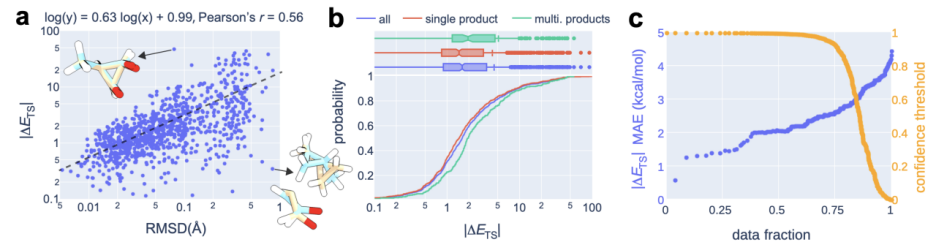 Le contenu qui doit être réécrit est : Illustration : OA- ReactDiff plus préconise la performance énergétique des structures TS. (Source : Paper)
Le contenu qui doit être réécrit est : Illustration : OA- ReactDiff plus préconise la performance énergétique des structures TS. (Source : Paper)
Ce qui doit être réécrit est : Cette recherche est la première à utiliser un modèle de diffusion 3D dans les réactions chimiques. L'importance de ce travail ne peut être ignorée, bien que les chercheurs n'aient étudié que des composés comportant un plus petit nombre d'atomes (
Le professeur Kulik a souligné : « Même face à des systèmes plus grands ou même à des systèmes catalysés par des enzymes, il est toujours possible d'obtenir des informations sur les différentes manières dont les atomes sont les plus susceptibles de se réorganiser. »
Les chercheurs envisagent désormais d'étendre leur modèle en ajoutant d'autres composants, comme des catalyseurs. Tirant parti du caractère aléatoire de l’IA générative, OA-ReactDiff peut explorer des réactions chimiques inattendues. Cette fonctionnalité complète le cadre d'exploration intuitive des réactions basé sur la chimie, aide à établir un réseau de réactions chimiques plus complet et aide au développement et à la conception de nouveaux matériaux catalytiques. La recherche dans ce domaine peut les aider à accélérer la découverte de nouveaux catalyseurs pour des réactions spécifiques. De plus, l’algorithme proposé pourrait être utile pour développer de nouveaux procédés pour les médicaments, les carburants ou d’autres composés utiles, en particulier lorsque la synthèse implique de nombreuses étapes chimiques.
Le Dr Duan Chenru a souligné que dans le passé, tous ces calculs étaient effectués à l'aide de méthodes de chimie quantique, mais que nous pouvons désormais remplacer la chimie quantique par des modèles génératifs plus rapides.
Les chercheurs ont également souligné que les réactions chimiques sont au cœur de la recherche chimique. . En plus de la conception de catalyseurs orientée vers les applications industrielles, OA-ReactDiff a également de nombreuses applications potentielles intéressantes, telles que l'exploration des interactions gazeuses qui peuvent se produire sur d'autres planètes, la simulation de processus de réaction au cours de l'évolution des premiers stades de la vie sur Terre, etc.
Ce qui précède est le contenu détaillé de. pour plus d'informations, suivez d'autres articles connexes sur le site Web de PHP en chinois!

Outils d'IA chauds

Undresser.AI Undress
Application basée sur l'IA pour créer des photos de nu réalistes

AI Clothes Remover
Outil d'IA en ligne pour supprimer les vêtements des photos.

Undress AI Tool
Images de déshabillage gratuites

Clothoff.io
Dissolvant de vêtements AI

AI Hentai Generator
Générez AI Hentai gratuitement.

Article chaud
Outils chauds

Bloc-notes++7.3.1
Éditeur de code facile à utiliser et gratuit

SublimeText3 version chinoise
Version chinoise, très simple à utiliser

Envoyer Studio 13.0.1
Puissant environnement de développement intégré PHP

Dreamweaver CS6
Outils de développement Web visuel

SublimeText3 version Mac
Logiciel d'édition de code au niveau de Dieu (SublimeText3)



 Repoussant les limites de la détection de défauts traditionnelle, « Defect Spectrum » permet pour la première fois une détection de défauts industriels d'une ultra haute précision et d'une sémantique riche.
Jul 26, 2024 pm 05:38 PM
Repoussant les limites de la détection de défauts traditionnelle, « Defect Spectrum » permet pour la première fois une détection de défauts industriels d'une ultra haute précision et d'une sémantique riche.
Jul 26, 2024 pm 05:38 PM
Dans la fabrication moderne, une détection précise des défauts est non seulement la clé pour garantir la qualité des produits, mais également la clé de l’amélioration de l’efficacité de la production. Cependant, les ensembles de données de détection de défauts existants manquent souvent de précision et de richesse sémantique requises pour les applications pratiques, ce qui rend les modèles incapables d'identifier des catégories ou des emplacements de défauts spécifiques. Afin de résoudre ce problème, une équipe de recherche de premier plan composée de l'Université des sciences et technologies de Hong Kong, Guangzhou et de Simou Technology a développé de manière innovante l'ensemble de données « DefectSpectrum », qui fournit une annotation à grande échelle détaillée et sémantiquement riche des défauts industriels. Comme le montre le tableau 1, par rapport à d'autres ensembles de données industrielles, l'ensemble de données « DefectSpectrum » fournit le plus grand nombre d'annotations de défauts (5 438 échantillons de défauts) et la classification de défauts la plus détaillée (125 catégories de défauts).
 Le modèle de dialogue NVIDIA ChatQA a évolué vers la version 2.0, avec la longueur du contexte mentionnée à 128 Ko
Jul 26, 2024 am 08:40 AM
Le modèle de dialogue NVIDIA ChatQA a évolué vers la version 2.0, avec la longueur du contexte mentionnée à 128 Ko
Jul 26, 2024 am 08:40 AM
La communauté ouverte LLM est une époque où une centaine de fleurs fleurissent et s'affrontent. Vous pouvez voir Llama-3-70B-Instruct, QWen2-72B-Instruct, Nemotron-4-340B-Instruct, Mixtral-8x22BInstruct-v0.1 et bien d'autres. excellents interprètes. Cependant, par rapport aux grands modèles propriétaires représentés par le GPT-4-Turbo, les modèles ouverts présentent encore des lacunes importantes dans de nombreux domaines. En plus des modèles généraux, certains modèles ouverts spécialisés dans des domaines clés ont été développés, tels que DeepSeek-Coder-V2 pour la programmation et les mathématiques, et InternVL pour les tâches de langage visuel.
 Google AI a remporté la médaille d'argent de l'Olympiade mathématique de l'OMI, le modèle de raisonnement mathématique AlphaProof a été lancé et l'apprentissage par renforcement est de retour.
Jul 26, 2024 pm 02:40 PM
Google AI a remporté la médaille d'argent de l'Olympiade mathématique de l'OMI, le modèle de raisonnement mathématique AlphaProof a été lancé et l'apprentissage par renforcement est de retour.
Jul 26, 2024 pm 02:40 PM
Pour l’IA, l’Olympiade mathématique n’est plus un problème. Jeudi, l'intelligence artificielle de Google DeepMind a réalisé un exploit : utiliser l'IA pour résoudre la vraie question de l'Olympiade mathématique internationale de cette année, l'OMI, et elle n'était qu'à un pas de remporter la médaille d'or. Le concours de l'OMI qui vient de se terminer la semaine dernière comportait six questions portant sur l'algèbre, la combinatoire, la géométrie et la théorie des nombres. Le système d'IA hybride proposé par Google a répondu correctement à quatre questions et a marqué 28 points, atteignant le niveau de la médaille d'argent. Plus tôt ce mois-ci, le professeur titulaire de l'UCLA, Terence Tao, venait de promouvoir l'Olympiade mathématique de l'IA (AIMO Progress Award) avec un prix d'un million de dollars. De manière inattendue, le niveau de résolution de problèmes d'IA s'était amélioré à ce niveau avant juillet. Posez les questions simultanément sur l'OMI. La chose la plus difficile à faire correctement est l'OMI, qui a la plus longue histoire, la plus grande échelle et la plus négative.
 Le point de vue de la nature : les tests de l'intelligence artificielle en médecine sont dans le chaos. Que faut-il faire ?
Aug 22, 2024 pm 04:37 PM
Le point de vue de la nature : les tests de l'intelligence artificielle en médecine sont dans le chaos. Que faut-il faire ?
Aug 22, 2024 pm 04:37 PM
Editeur | ScienceAI Sur la base de données cliniques limitées, des centaines d'algorithmes médicaux ont été approuvés. Les scientifiques se demandent qui devrait tester les outils et comment le faire au mieux. Devin Singh a vu un patient pédiatrique aux urgences subir un arrêt cardiaque alors qu'il attendait un traitement pendant une longue période, ce qui l'a incité à explorer l'application de l'IA pour réduire les temps d'attente. À l’aide des données de triage des salles d’urgence de SickKids, Singh et ses collègues ont construit une série de modèles d’IA pour fournir des diagnostics potentiels et recommander des tests. Une étude a montré que ces modèles peuvent accélérer les visites chez le médecin de 22,3 %, accélérant ainsi le traitement des résultats de près de 3 heures par patient nécessitant un examen médical. Cependant, le succès des algorithmes d’intelligence artificielle dans la recherche ne fait que le vérifier.
 Formation avec des millions de données cristallines pour résoudre le problème de la phase cristallographique, la méthode d'apprentissage profond PhAI est publiée dans Science
Aug 08, 2024 pm 09:22 PM
Formation avec des millions de données cristallines pour résoudre le problème de la phase cristallographique, la méthode d'apprentissage profond PhAI est publiée dans Science
Aug 08, 2024 pm 09:22 PM
Editeur | KX À ce jour, les détails structurels et la précision déterminés par cristallographie, des métaux simples aux grandes protéines membranaires, sont inégalés par aucune autre méthode. Cependant, le plus grand défi, appelé problème de phase, reste la récupération des informations de phase à partir d'amplitudes déterminées expérimentalement. Des chercheurs de l'Université de Copenhague au Danemark ont développé une méthode d'apprentissage en profondeur appelée PhAI pour résoudre les problèmes de phase cristalline. Un réseau neuronal d'apprentissage en profondeur formé à l'aide de millions de structures cristallines artificielles et de leurs données de diffraction synthétique correspondantes peut générer des cartes précises de densité électronique. L'étude montre que cette méthode de solution structurelle ab initio basée sur l'apprentissage profond peut résoudre le problème de phase avec une résolution de seulement 2 Angströms, ce qui équivaut à seulement 10 à 20 % des données disponibles à la résolution atomique, alors que le calcul ab initio traditionnel
 Afin de fournir un nouveau système de référence et d'évaluation de questions-réponses scientifiques et complexes pour les grands modèles, l'UNSW, Argonne, l'Université de Chicago et d'autres institutions ont lancé conjointement le cadre SciQAG.
Jul 25, 2024 am 06:42 AM
Afin de fournir un nouveau système de référence et d'évaluation de questions-réponses scientifiques et complexes pour les grands modèles, l'UNSW, Argonne, l'Université de Chicago et d'autres institutions ont lancé conjointement le cadre SciQAG.
Jul 25, 2024 am 06:42 AM
L'ensemble de données ScienceAI Question Answering (QA) joue un rôle essentiel dans la promotion de la recherche sur le traitement du langage naturel (NLP). Des ensembles de données d'assurance qualité de haute qualité peuvent non seulement être utilisés pour affiner les modèles, mais également évaluer efficacement les capacités des grands modèles linguistiques (LLM), en particulier la capacité à comprendre et à raisonner sur les connaissances scientifiques. Bien qu’il existe actuellement de nombreux ensembles de données scientifiques d’assurance qualité couvrant la médecine, la chimie, la biologie et d’autres domaines, ces ensembles de données présentent encore certaines lacunes. Premièrement, le formulaire de données est relativement simple, et la plupart sont des questions à choix multiples. Elles sont faciles à évaluer, mais limitent la plage de sélection des réponses du modèle et ne peuvent pas tester pleinement la capacité du modèle à répondre aux questions scientifiques. En revanche, les questions et réponses ouvertes
 Identifiez automatiquement les meilleures molécules et réduisez les coûts de synthèse. Le MIT développe un cadre d'algorithme de prise de décision en matière de conception moléculaire.
Jun 22, 2024 am 06:43 AM
Identifiez automatiquement les meilleures molécules et réduisez les coûts de synthèse. Le MIT développe un cadre d'algorithme de prise de décision en matière de conception moléculaire.
Jun 22, 2024 am 06:43 AM
Éditeur | L’utilisation de Ziluo AI pour rationaliser la découverte de médicaments explose. Ciblez des milliards de molécules candidates pour détecter celles qui pourraient posséder les propriétés nécessaires au développement de nouveaux médicaments. Il y a tellement de variables à prendre en compte, depuis le prix des matériaux jusqu’au risque d’erreur, qu’évaluer les coûts de synthèse des meilleures molécules candidates n’est pas une tâche facile, même si les scientifiques utilisent l’IA. Ici, les chercheurs du MIT ont développé SPARROW, un cadre d'algorithme de prise de décision quantitative, pour identifier automatiquement les meilleurs candidats moléculaires, minimisant ainsi les coûts de synthèse tout en maximisant la probabilité que les candidats possèdent les propriétés souhaitées. L’algorithme a également identifié les matériaux et les étapes expérimentales nécessaires à la synthèse de ces molécules. SPARROW prend en compte le coût de synthèse d'un lot de molécules à la fois, puisque plusieurs molécules candidates sont souvent disponibles
 Les performances de SOTA, la méthode d'IA de prédiction d'affinité protéine-ligand multimodale de Xiamen, combinent pour la première fois des informations sur la surface moléculaire
Jul 17, 2024 pm 06:37 PM
Les performances de SOTA, la méthode d'IA de prédiction d'affinité protéine-ligand multimodale de Xiamen, combinent pour la première fois des informations sur la surface moléculaire
Jul 17, 2024 pm 06:37 PM
Editeur | KX Dans le domaine de la recherche et du développement de médicaments, il est crucial de prédire avec précision et efficacité l'affinité de liaison des protéines et des ligands pour le criblage et l'optimisation des médicaments. Cependant, les études actuelles ne prennent pas en compte le rôle important des informations sur la surface moléculaire dans les interactions protéine-ligand. Sur cette base, des chercheurs de l'Université de Xiamen ont proposé un nouveau cadre d'extraction de caractéristiques multimodales (MFE), qui combine pour la première fois des informations sur la surface des protéines, la structure et la séquence 3D, et utilise un mécanisme d'attention croisée pour comparer différentes modalités. alignement. Les résultats expérimentaux démontrent que cette méthode atteint des performances de pointe dans la prédiction des affinités de liaison protéine-ligand. De plus, les études d’ablation démontrent l’efficacité et la nécessité des informations sur la surface des protéines et de l’alignement des caractéristiques multimodales dans ce cadre. Les recherches connexes commencent par "S
