
 Périphériques technologiques
IA
Précision > 98 %, GPT basé sur la densité électronique est utilisé dans la recherche chimique, publié dans la sous-journal Nature
Périphériques technologiques
IA
Précision > 98 %, GPT basé sur la densité électronique est utilisé dans la recherche chimique, publié dans la sous-journal Nature
Précision > 98 %, GPT basé sur la densité électronique est utilisé dans la recherche chimique, publié dans la sous-journal Nature

Editeur | Violet
L'espace chimique des molécules synthétisées est très large. Une exploration efficace de ce domaine nécessite la confiance sur le criblage informatique Des technologies, telles que l'apprentissage profond, pour découvrir rapidement une variété de composés intéressants
La conversion des structures moléculaires en représentations numériques et le développement d'algorithmes correspondants pour générer de nouvelles structures moléculaires sont la clé de la découverte chimique
Récemment, l'Université de Glasgow, Royaume-Uni L'équipe de recherche a proposé un modèle d'apprentissage automatique basé sur la formation à la densité électronique pour générer des classeurs hôte-invité. Ce modèle est capable de lire des données au format SMILES (Simplified Molecular Linear Input Specification) avec une précision allant jusqu'à 98 %. une précision de 98 % Description complète des molécules dans un espace bidimensionnel
Générez une représentation tridimensionnelle de la densité électronique et du potentiel électrostatique du système hôte-invité via un auto-encodeur variationnel, puis optimisez la génération de l'invité. par descente de gradient. Enfin, utilisez le transformateur pour convertir l'invité en SMILES. Une représentation et une transformation efficaces des structures invitées ont été appliquées avec succès aux systèmes hôtes moléculaires établis, au cucurbituril et aux cages métallo-organiques, ce qui a permis la découverte de 9. invités CB[6] précédemment validés et 7 objets non signalés, et découvert 4 GPT à 98 % non signalés basés sur la densité électronique pour la recherche chimique, publiés dans la sous-journal Nature " />Objets.
La recherche s'intitulait « GPT basé sur la densité électronique pour l'optimisation et la suggestion de classeurs hôte-invité » et a été publiée dans « Nature Computational Science » le 8 mars 2024. 
Lien papier : https://www.nature.com/articles/s43588-024-00602-x 
Un modèle d'apprentissage automatique formé sur la densité électronique
Ici, il est démontré que la représentation d'une molécule hôte sous la forme d'un volume 3D (c'est-à-dire une densité électronique modifiée avec un potentiel électrostatique) permet une découverte assistée par ordinateur des invités de l'hôte. pas besoin de comprendre le système hôte-invité au-delà de la structure chimique du sujet.
Au cours du processus, les chercheurs ont construit un modèle Transformer qui peut être entraîné pour convertir efficacement des descripteurs moléculaires volumétriques 3D en représentations SMILES, générant ainsi des structures moléculaires utilisables par les chimistes professionnels. L'étude a également révélé que les molécules peuvent être représentées efficacement sous forme de volumes 3D en modifiant leur densité électronique avec des données de potentiel électrostatique, et que ces deux caractéristiques sont suffisantes pour optimiser la forme et la charge du volume entre les descripteurs 3D en utilisant un schéma d'échantillonnage autorégressif. Molécules invitées interagissez pour découvrir l’hôte.98 %, le GPT basé sur la densité électronique est utilisé dans la recherche chimique, publié dans le sous-journal Nature" />
![准确率 ></section>Le modèle Transformer prédit parfaitement sa représentation SMILES avec une précision de 98,125 %. La précision de prédiction pour un seul jeton est de 99,114 %. modèle génératif <section></section> <p>Aperçu du workflow</p><p>La découverte assistée par ordinateur du cucurbituril CB[6] et la validation expérimentale des cages métallo-organiques <p> nécessitent un flux de travail à deux niveaux. Tout d’abord, un flux de travail in silico a été conçu pour générer des bibliothèques virtuelles de molécules invitées potentielles pour les deux hôtes. Un flux de travail in vitro a ensuite été établi, comprenant la sélection des candidats invités les plus prometteurs de ces bibliothèques virtuelles par des chimistes experts pour des tests expérimentaux. <img src=](https://img.php.cn/upload/article/000/000/164/171152017578536.png)
98 %, le GPT basé sur la densité électronique est utilisé dans la recherche chimique, publié dans le sous-journal Nature" />![准确率 ></p>Illustration : Découverte de nouvelles molécules invitées grâce à la représentation volumique de la densité électronique. (Source : article) <section></section>CB[ 6] et <p> La génération informatique de molécules invitées est réalisée grâce au flux de travail illustré dans la figure ci-dessus, <img src=](https://img.php.cn/upload/article/000/000/164/171152017597952.png) Le flux de travail comprend les étapes suivantes :
Le flux de travail comprend les étapes suivantes :
grâce à des simulations informatiques, un flux de travail in vitro a été établi pour tester expérimentalement les candidats les plus prometteurs. 
générés grâce à son flux de travail informatique ont été classés par des experts en chimie pour des tests expérimentaux. Les invités prometteurs à tester sont sélectionnés en fonction de leur similarité structurelle avec les invités connus de CB[6] ou , de l'intuition des chimistes professionnels et de leur disponibilité commerciale.
(2) Utilisation directe du  Méthode de titrage pour déterminer l'affinité du CB[6] ou . Il est à noter que l'invité généré dans l'ordinateur Mélanges contenant des molécules connues auparavant pour se lier à (ou étroitement liées) à des hôtes et des molécules qui défient l'intuition des experts
Méthode de titrage pour déterminer l'affinité du CB[6] ou . Il est à noter que l'invité généré dans l'ordinateur Mélanges contenant des molécules connues auparavant pour se lier à (ou étroitement liées) à des hôtes et des molécules qui défient l'intuition des experts
Validation expérimentale de deux systèmes hôte-invité communs
Les chercheurs ont validé expérimentalement leur flux de travail pour deux hôtes communs. -systèmes invités : le cucurbituril (CB[n]) et les cages métallo-organiques sont devenus des objets vérifiés dans la littérature et non signalés. L'algorithme a généré 9 objets précédemment connus pour CB[6]. Sept nouveaux invités potentiels de CB[6] ont également été identifiés. et l'affinité de CB[6] pour ces nouveaux invités a été évaluée par
titrage direct en HCO2H/H2O 1:1v/vDans les 7 cas, un ensemble de signaux est observé pour le système hôte-invité, indiquant que le système hôte-invité. Le système subit un échange rapide sur l'échelle de temps RMN. Après la complexation, la résonance de la chaîne aliphatique des molécules invitées se déplace vers le haut, indiquant qu'elles sont encapsulées dans la cavité CB[6] trouvée  98 %, GPT basé sur la densité électronique. Chemical Research, publié dans Nature Sub-Journal" /> La constante d'association avec CB[6] suit la tendance précédemment établie, allant de 13,5 M^− 1 à 5 470 M^−1.
98 %, GPT basé sur la densité électronique. Chemical Research, publié dans Nature Sub-Journal" /> La constante d'association avec CB[6] suit la tendance précédemment établie, allant de 13,5 M^− 1 à 5 470 M^−1.

Illustration : optimisation de CB[6] et objets précédemment connus et objets optimisés de 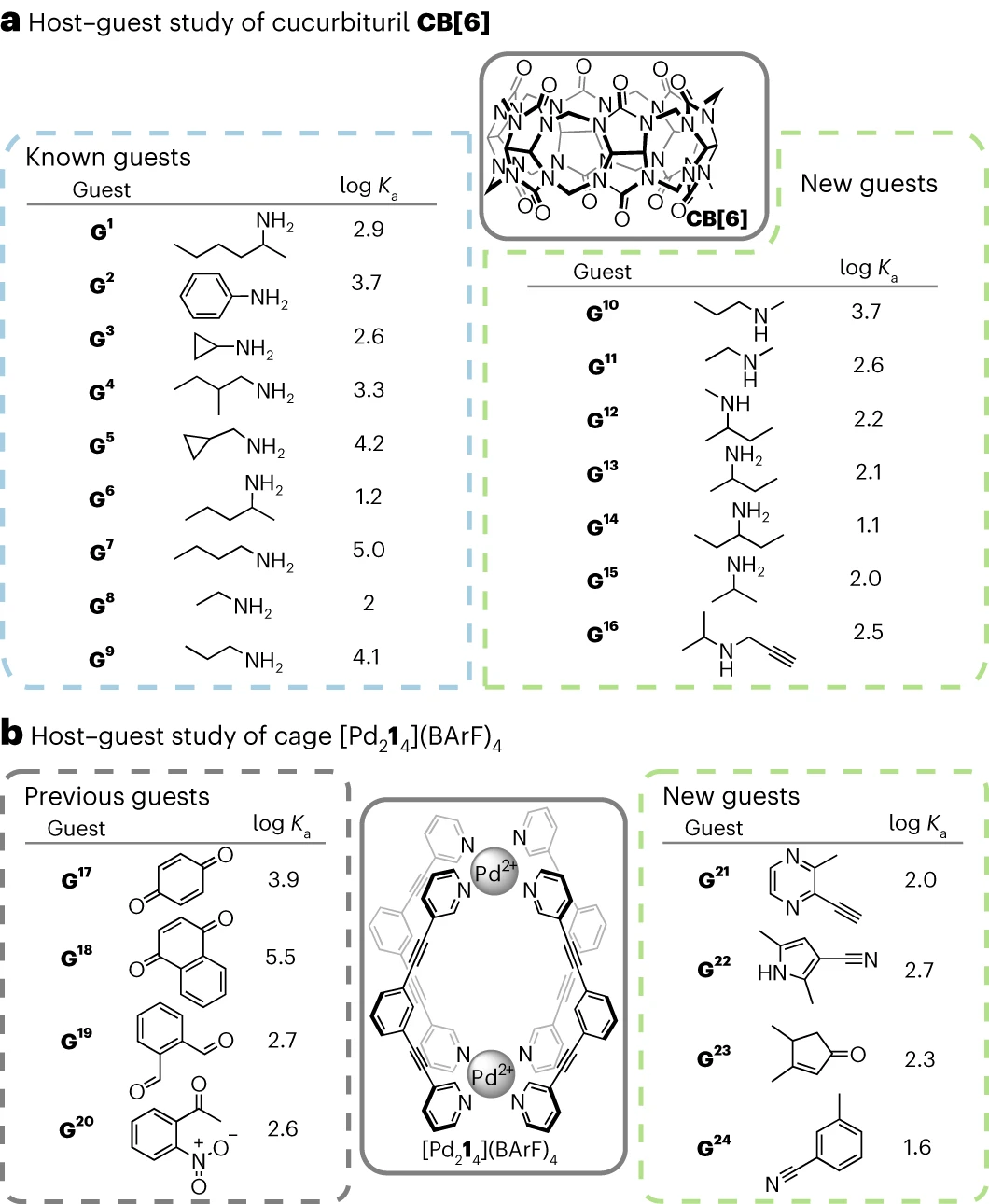 , l'algorithme d'optimisation a généré uniquement des molécules invitées inconnues, et la force de liaison entre quatre invités potentiels non signalés et [Pd214](BArF)4 a été testée par titrage direct
, l'algorithme d'optimisation a généré uniquement des molécules invitées inconnues, et la force de liaison entre quatre invités potentiels non signalés et [Pd214](BArF)4 a été testée par titrage direct
Alors que la recherche s'est concentrée sur l'utilisation de la notation SMILES pour représenter des molécules, d'autres formats similaires tels que les chaînes intégrées auto-référentielles (SELFIES) ont également été testés.
Bien que l'ensemble de données QM9 contienne des molécules parfaitement dimensionnées pour devenir des invités pour des hôtes tels que CB[6], une limitation rencontrée par cette étude est que les cages métallo-organiques ont des cavités plus grandes et nécessitent des molécules invitées plus grosses. Dans les études futures, des ensembles de données contenant des molécules plus grosses, tels que l'ensemble de données GDB-17, seront utilisés.
Après cela, « Notre objectif est d'intégrer la sélection de nouveaux ligands dans le processus de génération, de synthétiser de manière autonome des molécules sur des plateformes de synthèse automatisées (telles que les robots Chemputer), de boucler la boucle entre optimisation et tests et de créer un environnement cyber-physique fermé. Système de boucle."
Ce qui précède est le contenu détaillé de. pour plus d'informations, suivez d'autres articles connexes sur le site Web de PHP en chinois!

Outils d'IA chauds

Undresser.AI Undress
Application basée sur l'IA pour créer des photos de nu réalistes

AI Clothes Remover
Outil d'IA en ligne pour supprimer les vêtements des photos.

Undress AI Tool
Images de déshabillage gratuites

Clothoff.io
Dissolvant de vêtements AI

AI Hentai Generator
Générez AI Hentai gratuitement.

Article chaud
Outils chauds

Bloc-notes++7.3.1
Éditeur de code facile à utiliser et gratuit

SublimeText3 version chinoise
Version chinoise, très simple à utiliser

Envoyer Studio 13.0.1
Puissant environnement de développement intégré PHP

Dreamweaver CS6
Outils de développement Web visuel

SublimeText3 version Mac
Logiciel d'édition de code au niveau de Dieu (SublimeText3)



 Repoussant les limites de la détection de défauts traditionnelle, « Defect Spectrum » permet pour la première fois une détection de défauts industriels d'une ultra haute précision et d'une sémantique riche.
Jul 26, 2024 pm 05:38 PM
Repoussant les limites de la détection de défauts traditionnelle, « Defect Spectrum » permet pour la première fois une détection de défauts industriels d'une ultra haute précision et d'une sémantique riche.
Jul 26, 2024 pm 05:38 PM
Dans la fabrication moderne, une détection précise des défauts est non seulement la clé pour garantir la qualité des produits, mais également la clé de l’amélioration de l’efficacité de la production. Cependant, les ensembles de données de détection de défauts existants manquent souvent de précision et de richesse sémantique requises pour les applications pratiques, ce qui rend les modèles incapables d'identifier des catégories ou des emplacements de défauts spécifiques. Afin de résoudre ce problème, une équipe de recherche de premier plan composée de l'Université des sciences et technologies de Hong Kong, Guangzhou et de Simou Technology a développé de manière innovante l'ensemble de données « DefectSpectrum », qui fournit une annotation à grande échelle détaillée et sémantiquement riche des défauts industriels. Comme le montre le tableau 1, par rapport à d'autres ensembles de données industrielles, l'ensemble de données « DefectSpectrum » fournit le plus grand nombre d'annotations de défauts (5 438 échantillons de défauts) et la classification de défauts la plus détaillée (125 catégories de défauts).
 Le modèle de dialogue NVIDIA ChatQA a évolué vers la version 2.0, avec la longueur du contexte mentionnée à 128 Ko
Jul 26, 2024 am 08:40 AM
Le modèle de dialogue NVIDIA ChatQA a évolué vers la version 2.0, avec la longueur du contexte mentionnée à 128 Ko
Jul 26, 2024 am 08:40 AM
La communauté ouverte LLM est une époque où une centaine de fleurs fleurissent et s'affrontent. Vous pouvez voir Llama-3-70B-Instruct, QWen2-72B-Instruct, Nemotron-4-340B-Instruct, Mixtral-8x22BInstruct-v0.1 et bien d'autres. excellents interprètes. Cependant, par rapport aux grands modèles propriétaires représentés par le GPT-4-Turbo, les modèles ouverts présentent encore des lacunes importantes dans de nombreux domaines. En plus des modèles généraux, certains modèles ouverts spécialisés dans des domaines clés ont été développés, tels que DeepSeek-Coder-V2 pour la programmation et les mathématiques, et InternVL pour les tâches de langage visuel.
 Google AI a remporté la médaille d'argent de l'Olympiade mathématique de l'OMI, le modèle de raisonnement mathématique AlphaProof a été lancé et l'apprentissage par renforcement est de retour.
Jul 26, 2024 pm 02:40 PM
Google AI a remporté la médaille d'argent de l'Olympiade mathématique de l'OMI, le modèle de raisonnement mathématique AlphaProof a été lancé et l'apprentissage par renforcement est de retour.
Jul 26, 2024 pm 02:40 PM
Pour l’IA, l’Olympiade mathématique n’est plus un problème. Jeudi, l'intelligence artificielle de Google DeepMind a réalisé un exploit : utiliser l'IA pour résoudre la vraie question de l'Olympiade mathématique internationale de cette année, l'OMI, et elle n'était qu'à un pas de remporter la médaille d'or. Le concours de l'OMI qui vient de se terminer la semaine dernière comportait six questions portant sur l'algèbre, la combinatoire, la géométrie et la théorie des nombres. Le système d'IA hybride proposé par Google a répondu correctement à quatre questions et a marqué 28 points, atteignant le niveau de la médaille d'argent. Plus tôt ce mois-ci, le professeur titulaire de l'UCLA, Terence Tao, venait de promouvoir l'Olympiade mathématique de l'IA (AIMO Progress Award) avec un prix d'un million de dollars. De manière inattendue, le niveau de résolution de problèmes d'IA s'était amélioré à ce niveau avant juillet. Posez les questions simultanément sur l'OMI. La chose la plus difficile à faire correctement est l'OMI, qui a la plus longue histoire, la plus grande échelle et la plus négative.
 Le point de vue de la nature : les tests de l'intelligence artificielle en médecine sont dans le chaos. Que faut-il faire ?
Aug 22, 2024 pm 04:37 PM
Le point de vue de la nature : les tests de l'intelligence artificielle en médecine sont dans le chaos. Que faut-il faire ?
Aug 22, 2024 pm 04:37 PM
Editeur | ScienceAI Sur la base de données cliniques limitées, des centaines d'algorithmes médicaux ont été approuvés. Les scientifiques se demandent qui devrait tester les outils et comment le faire au mieux. Devin Singh a vu un patient pédiatrique aux urgences subir un arrêt cardiaque alors qu'il attendait un traitement pendant une longue période, ce qui l'a incité à explorer l'application de l'IA pour réduire les temps d'attente. À l’aide des données de triage des salles d’urgence de SickKids, Singh et ses collègues ont construit une série de modèles d’IA pour fournir des diagnostics potentiels et recommander des tests. Une étude a montré que ces modèles peuvent accélérer les visites chez le médecin de 22,3 %, accélérant ainsi le traitement des résultats de près de 3 heures par patient nécessitant un examen médical. Cependant, le succès des algorithmes d’intelligence artificielle dans la recherche ne fait que le vérifier.
 Formation avec des millions de données cristallines pour résoudre le problème de la phase cristallographique, la méthode d'apprentissage profond PhAI est publiée dans Science
Aug 08, 2024 pm 09:22 PM
Formation avec des millions de données cristallines pour résoudre le problème de la phase cristallographique, la méthode d'apprentissage profond PhAI est publiée dans Science
Aug 08, 2024 pm 09:22 PM
Editeur | KX À ce jour, les détails structurels et la précision déterminés par cristallographie, des métaux simples aux grandes protéines membranaires, sont inégalés par aucune autre méthode. Cependant, le plus grand défi, appelé problème de phase, reste la récupération des informations de phase à partir d'amplitudes déterminées expérimentalement. Des chercheurs de l'Université de Copenhague au Danemark ont développé une méthode d'apprentissage en profondeur appelée PhAI pour résoudre les problèmes de phase cristalline. Un réseau neuronal d'apprentissage en profondeur formé à l'aide de millions de structures cristallines artificielles et de leurs données de diffraction synthétique correspondantes peut générer des cartes précises de densité électronique. L'étude montre que cette méthode de solution structurelle ab initio basée sur l'apprentissage profond peut résoudre le problème de phase avec une résolution de seulement 2 Angströms, ce qui équivaut à seulement 10 à 20 % des données disponibles à la résolution atomique, alors que le calcul ab initio traditionnel
 Afin de fournir un nouveau système de référence et d'évaluation de questions-réponses scientifiques et complexes pour les grands modèles, l'UNSW, Argonne, l'Université de Chicago et d'autres institutions ont lancé conjointement le cadre SciQAG.
Jul 25, 2024 am 06:42 AM
Afin de fournir un nouveau système de référence et d'évaluation de questions-réponses scientifiques et complexes pour les grands modèles, l'UNSW, Argonne, l'Université de Chicago et d'autres institutions ont lancé conjointement le cadre SciQAG.
Jul 25, 2024 am 06:42 AM
L'ensemble de données ScienceAI Question Answering (QA) joue un rôle essentiel dans la promotion de la recherche sur le traitement du langage naturel (NLP). Des ensembles de données d'assurance qualité de haute qualité peuvent non seulement être utilisés pour affiner les modèles, mais également évaluer efficacement les capacités des grands modèles linguistiques (LLM), en particulier la capacité à comprendre et à raisonner sur les connaissances scientifiques. Bien qu’il existe actuellement de nombreux ensembles de données scientifiques d’assurance qualité couvrant la médecine, la chimie, la biologie et d’autres domaines, ces ensembles de données présentent encore certaines lacunes. Premièrement, le formulaire de données est relativement simple, et la plupart sont des questions à choix multiples. Elles sont faciles à évaluer, mais limitent la plage de sélection des réponses du modèle et ne peuvent pas tester pleinement la capacité du modèle à répondre aux questions scientifiques. En revanche, les questions et réponses ouvertes
 Identifiez automatiquement les meilleures molécules et réduisez les coûts de synthèse. Le MIT développe un cadre d'algorithme de prise de décision en matière de conception moléculaire.
Jun 22, 2024 am 06:43 AM
Identifiez automatiquement les meilleures molécules et réduisez les coûts de synthèse. Le MIT développe un cadre d'algorithme de prise de décision en matière de conception moléculaire.
Jun 22, 2024 am 06:43 AM
Éditeur | L’utilisation de Ziluo AI pour rationaliser la découverte de médicaments explose. Ciblez des milliards de molécules candidates pour détecter celles qui pourraient posséder les propriétés nécessaires au développement de nouveaux médicaments. Il y a tellement de variables à prendre en compte, depuis le prix des matériaux jusqu’au risque d’erreur, qu’évaluer les coûts de synthèse des meilleures molécules candidates n’est pas une tâche facile, même si les scientifiques utilisent l’IA. Ici, les chercheurs du MIT ont développé SPARROW, un cadre d'algorithme de prise de décision quantitative, pour identifier automatiquement les meilleurs candidats moléculaires, minimisant ainsi les coûts de synthèse tout en maximisant la probabilité que les candidats possèdent les propriétés souhaitées. L’algorithme a également identifié les matériaux et les étapes expérimentales nécessaires à la synthèse de ces molécules. SPARROW prend en compte le coût de synthèse d'un lot de molécules à la fois, puisque plusieurs molécules candidates sont souvent disponibles
 Les performances de SOTA, la méthode d'IA de prédiction d'affinité protéine-ligand multimodale de Xiamen, combinent pour la première fois des informations sur la surface moléculaire
Jul 17, 2024 pm 06:37 PM
Les performances de SOTA, la méthode d'IA de prédiction d'affinité protéine-ligand multimodale de Xiamen, combinent pour la première fois des informations sur la surface moléculaire
Jul 17, 2024 pm 06:37 PM
Editeur | KX Dans le domaine de la recherche et du développement de médicaments, il est crucial de prédire avec précision et efficacité l'affinité de liaison des protéines et des ligands pour le criblage et l'optimisation des médicaments. Cependant, les études actuelles ne prennent pas en compte le rôle important des informations sur la surface moléculaire dans les interactions protéine-ligand. Sur cette base, des chercheurs de l'Université de Xiamen ont proposé un nouveau cadre d'extraction de caractéristiques multimodales (MFE), qui combine pour la première fois des informations sur la surface des protéines, la structure et la séquence 3D, et utilise un mécanisme d'attention croisée pour comparer différentes modalités. alignement. Les résultats expérimentaux démontrent que cette méthode atteint des performances de pointe dans la prédiction des affinités de liaison protéine-ligand. De plus, les études d’ablation démontrent l’efficacité et la nécessité des informations sur la surface des protéines et de l’alignement des caractéristiques multimodales dans ce cadre. Les recherches connexes commencent par "S
