

機械学習モデルが有機反応機構を優れた精度で分類

化学反応の発見は、実験データをいかに早く取得できるかだけでなく、化学者がそのデータをいかに理解しやすいかによっても影響されます。新しい触媒反応の機構的基礎を明らかにすることは特に複雑な問題であり、多くの場合、計算および物理有機化学の専門知識が必要です。ただし、触媒反応は最も効率的な化学プロセスであるため、触媒反応を研究することが重要です。
最近、英国のマンチェスター大学 (UoM) 化学科の Burés 氏と Larrosa 氏は、ディープ ニューラル ネットワーク モデルが次のようなことができることを実証した機械学習モデルを報告しました。ユーザーによる追加入力なしで、通常のダイナミクス学習データを分析し、対応する機構カテゴリを自動的に解明するようにトレーニングを受けることができます。このモデルはさまざまなタイプのメカニズムを優れた精度で識別します。
今回の調査結果は、AI によるメカニズム分類がメカニズムの解明を簡素化および自動化できる強力な新しいツールであることを示しています。この研究により、完全に自動化された有機反応の発見と開発がさらに前進すると期待されています。
研究のタイトルは「機械学習を用いた有機反応機構の分類」で、2023年1月25日の「Nature」に掲載されました。 。

論文リンク: https://www.nature.com/articles/s41586 - 022-05639-4
化学反応のメカニズムを解明する伝統的な方法
製品の製造に必要な基本ステップの正確な順序は、合成方法を合理的に改善し、新しい触媒を設計し、工業プロセスを安全にスケールアップするために重要です。反応メカニズムを解明するには、複数の反応速度曲線を収集する必要があり、人間の専門家がデータに対して速度論解析を実行する必要があります。反応モニタリング技術は過去数十年にわたって、速度論的データ収集を完全に自動化できるまでに大幅に進歩しましたが、機構解明の基礎となる理論的枠組みは同じペースで進化していません。
現在の速度論解析パイプラインは、実験データから速度論的特性を抽出すること、考えられるすべてのメカニズムの速度論的特性を予測すること、実験的に抽出された特性を比較のために予測特性と組み合わせるという 3 つの主要なステップで構成されています。
1 世紀以上にわたり、化学者は反応速度から機構に関する情報を抽出してきました。現在でも使用されている方法の 1 つは、出発物質の最初の数パーセントの消費に焦点を当てて、反応の初速度を評価することです。この方法は、ほとんどの場合、時間の経過に伴う反応物濃度の変化が反応の開始時に線形であり、分析が簡単であるため、一般的です。この手法は洞察力に富んでいますが、時間の経過のほとんどにわたって発生する反応速度と濃度の変化を無視しています。
過去数十年にわたり、反応プロセス全体を通じて反応成分の濃度を評価するためのより高度な方法が開発されてきました。これらの方法は、反応速度図から反応ステップに関与する成分の数 (反応成分の順序とも呼ばれる) を明らかにする数学的手法によってさらに容易になります。これらの技術は確かに化学反応性についての洞察を提供し続けるでしょうが、触媒システムの反応速度論的挙動を説明するより包括的な機構仮説を提供するのではなく、反応成分の順序を分析することに限定されています。

図 1: 反応速度論解析の関連性と最先端の技術。 (出典: 論文)
AI は反応速度論分析の分野を変える
機械学習は化学者が問題を解決する方法に革命をもたらしています。分子と分子を合成するルートを設計し、反応メカニズムを理解します。 Burés と Larrosa は現在、機械学習モデルを使用して、シミュレートされた速度論的特性に基づいて反応を分類することにより、この革命を速度論的分析にもたらしています。
ここでは、研究者らは、シミュレートされた動力学データでトレーニングされた深層学習モデルが、時間的濃度分布からさまざまなメカニズムを正確に解明できることを実証しています。機械学習モデルは、速度則の導出や速度論的特性の抽出と予測の必要性を排除することで速度論的分析を簡素化し、すべての合成実験室での反応メカニズムの解明を大幅に促進します。
利用可能なすべての反応速度論データの総合的な分析により、このメソッドは反応曲線を調べる能力を向上させ、反応速度論分析中の潜在的な人的エラーを排除し、利用可能な反応速度論の範囲を拡大します。分析には、非定常状態 (活性化および非活性化プロセスを含む) および可逆反応が含まれます。このアプローチは、現在利用可能な速度論的分析方法を補完し、最も困難な状況で特に役立つでしょう。
具体的な研究
研究者らは、反応メカニズムの 20 カテゴリーを定義し、各カテゴリーの速度則を開発しました。各メカニズムは、一連の運動定数 (k1, … kn #) で構成されます。 ##) と化学物質の濃度は、常微分方程式 (ODE) 関数によって数学的に記述されます。次に、これらの方程式を解き、反応物の崩壊と生成物の生成を記述する何百万ものシミュレーションを生成しました。これらのシミュレートされた動力学データは、各機構クラスの特徴的な特徴を識別する学習アルゴリズムをトレーニングするために使用されます。結果として得られる分類モデルは、初期および時間濃度データを含む反応速度曲線を入力として使用し、反応の機構クラスを出力します。

# 図 2: メカニズムの範囲とデータ構成。 (出典: 論文)
深層学習モデルのトレーニングには大量のデータが必要になることが多く、このデータを実験的に収集する必要がある場合、大きな課題が生じる可能性があります。Burés と Larrosa のアルゴリズムのトレーニングへのアプローチは、大量の実験的運動データを生成するというボトルネックを回避します。この場合、研究者らは定常状態の近似を使用せずに、一連の ODE を数値的に解き、モデルのトレーニングと検証用に 500 万のダイナミクス サンプルを生成することができました。
モデルには 576,000 のトレーニング可能なパラメーターが含まれており、2 種類のニューラル ネットワークの組み合わせを使用します: (1) 長期および短期記憶ニューラル ネットワーク。時間データ シーケンスの処理に使用されます ( (2) 非時間データ (つまり、各反応速度論における触媒の初期濃度と長期短期記憶から抽出された特徴) を処理するための完全に接続されたニューラル ネットワーク。モデルは、合計が 1 になる各メカニズムの確率を出力します。
研究者らは、シミュレートされた運動曲線のテスト セットを使用してトレーニングされたモデルを評価し、これらの曲線が 92.6% の精度で機構クラスに正しく割り当てられていることを実証しました。

#このモデルは、「ノイズの多い」データが意図的に導入された場合でも良好に機能します。つまり、実験データの分類に使用できることを意味します。

最後に、研究者らは、以前に報告されたいくつかの実験的動力学曲線を使用してモデルのベンチマークを行いました。予測されたメカニズムは、以前の反応速度論的研究の結論とよく一致しています。場合によっては、モデルでは、元の作業では検出されなかった機構の詳細も特定されました。困難な反応に対して、モデルは 3 つの非常に類似した機構カテゴリーを提案します。しかし、著者らは、この結果はバグではなくモデルの特徴であると正しく述べており、メカニズムを解明するにはさらに具体的な実験が必要であることを示唆しています。 
図 5: 実験速度論データを使用したケーススタディ。 (出典: 論文)
要約すると、Burés と Larrosa は、速度論的研究から機構的仮説を導き出す長いプロセスを自動化するだけでなく、困難な物質の速度論的分析を実行する方法を開発しました。反応メカニズム。データ分析におけるあらゆる技術の進歩と同様に、結果として得られるメカニズムの分類は、さらなる実験的サポートを必要とする仮説としてみなされる必要があります。反応速度論データを誤って解釈するリスクは常に存在しますが、少数の実験に基づいて正しい反応経路を高精度で特定するアルゴリズムの能力により、より多くの研究者が反応速度論解析を試みるようになる可能性があります。
したがって、このアプローチは、特に化学者が機械学習アルゴリズムに精通するにつれて、反応開発プロセスへの速度論的分析の組み込みを普及させ、促進する可能性があります。
以上が機械学習モデルが有機反応機構を優れた精度で分類の詳細内容です。詳細については、PHP 中国語 Web サイトの他の関連記事を参照してください。

ホットAIツール

Undresser.AI Undress
リアルなヌード写真を作成する AI 搭載アプリ

AI Clothes Remover
写真から衣服を削除するオンライン AI ツール。

Undress AI Tool
脱衣画像を無料で

Clothoff.io
AI衣類リムーバー

Video Face Swap
完全無料の AI 顔交換ツールを使用して、あらゆるビデオの顔を簡単に交換できます。

人気の記事
ホットツール

メモ帳++7.3.1
使いやすく無料のコードエディター

SublimeText3 中国語版
中国語版、とても使いやすい

ゼンドスタジオ 13.0.1
強力な PHP 統合開発環境

ドリームウィーバー CS6
ビジュアル Web 開発ツール

SublimeText3 Mac版
神レベルのコード編集ソフト(SublimeText3)
ホットトピック
 7742
7742
 15
1643
14
1397
52
1291
25
1234
29
15
1643
14
1397
52
1291
25
1234
29

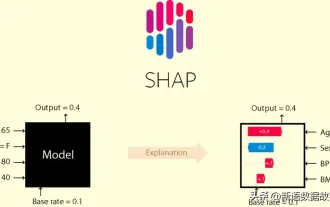 この記事では、SHAP: 機械学習のモデルの説明について説明します。
Jun 01, 2024 am 10:58 AM
この記事では、SHAP: 機械学習のモデルの説明について説明します。
Jun 01, 2024 am 10:58 AM
機械学習とデータ サイエンスの分野では、モデルの解釈可能性が常に研究者や実務家に焦点を当ててきました。深層学習やアンサンブル手法などの複雑なモデルが広く適用されるようになったことで、モデルの意思決定プロセスを理解することが特に重要になってきました。 Explainable AI|XAI は、モデルの透明性を高めることで、機械学習モデルに対する信頼と自信を構築するのに役立ちます。モデルの透明性の向上は、複数の複雑なモデルの普及や、モデルを説明するための意思決定プロセスなどの方法によって実現できます。これらの方法には、特徴重要度分析、モデル予測間隔推定、ローカル解釈可能性アルゴリズムなどが含まれます。特徴重要度分析では、入力特徴に対するモデルの影響度を評価することで、モデルの意思決定プロセスを説明できます。モデルの予測間隔の推定
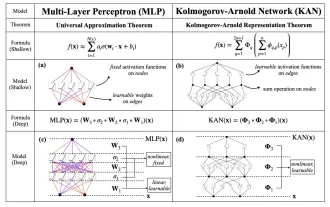 MLP に代わる KAN は、オープンソース プロジェクトによって畳み込みまで拡張されました
Jun 01, 2024 pm 10:03 PM
MLP に代わる KAN は、オープンソース プロジェクトによって畳み込みまで拡張されました
Jun 01, 2024 pm 10:03 PM
今月初め、MIT やその他の機関の研究者らは、MLP に代わる非常に有望な代替案である KAN を提案しました。 KAN は、精度と解釈可能性の点で MLP よりも優れています。また、非常に少数のパラメーターを使用して、多数のパラメーターを使用して実行する MLP よりも優れたパフォーマンスを発揮できます。たとえば、著者らは、KAN を使用して、より小規模なネットワークと高度な自動化で DeepMind の結果を再現したと述べています。具体的には、DeepMind の MLP には約 300,000 個のパラメーターがありますが、KAN には約 200 個のパラメーターしかありません。 KAN は、MLP が普遍近似定理に基づいているのに対し、KAN はコルモゴロフ-アーノルド表現定理に基づいているのと同様に、強力な数学的基礎を持っています。以下の図に示すように、KAN は
 C++ での機械学習アルゴリズムの実装: 一般的な課題と解決策
Jun 03, 2024 pm 01:25 PM
C++ での機械学習アルゴリズムの実装: 一般的な課題と解決策
Jun 03, 2024 pm 01:25 PM
C++ の機械学習アルゴリズムが直面する一般的な課題には、メモリ管理、マルチスレッド、パフォーマンスの最適化、保守性などがあります。解決策には、スマート ポインター、最新のスレッド ライブラリ、SIMD 命令、サードパーティ ライブラリの使用、コーディング スタイル ガイドラインの遵守、自動化ツールの使用が含まれます。実践的な事例では、Eigen ライブラリを使用して線形回帰アルゴリズムを実装し、メモリを効果的に管理し、高性能の行列演算を使用する方法を示します。
 総合的にDPOを超える:Chen Danqi氏のチームはシンプルなプリファレンス最適化SimPOを提案し、最強の8Bオープンソースモデルも洗練させた
Jun 01, 2024 pm 04:41 PM
総合的にDPOを超える:Chen Danqi氏のチームはシンプルなプリファレンス最適化SimPOを提案し、最強の8Bオープンソースモデルも洗練させた
Jun 01, 2024 pm 04:41 PM
大規模言語モデル (LLM) を人間の価値観や意図に合わせるには、人間のフィードバックを学習して、それが有用で、正直で、無害であることを確認することが重要です。 LLM を調整するという点では、ヒューマン フィードバックに基づく強化学習 (RLHF) が効果的な方法です。 RLHF 法の結果は優れていますが、最適化にはいくつかの課題があります。これには、報酬モデルをトレーニングし、その報酬を最大化するためにポリシー モデルを最適化することが含まれます。最近、一部の研究者はより単純なオフライン アルゴリズムを研究しており、その 1 つが直接優先最適化 (DPO) です。 DPO は、RLHF の報酬関数をパラメータ化することで、選好データに基づいてポリシー モデルを直接学習するため、明示的な報酬モデルの必要性がなくなります。この方法は簡単で安定しています
 あなたが知らない機械学習の 5 つの流派
Jun 05, 2024 pm 08:51 PM
あなたが知らない機械学習の 5 つの流派
Jun 05, 2024 pm 08:51 PM
機械学習は人工知能の重要な分野であり、明示的にプログラムしなくてもコンピューターにデータから学習して能力を向上させる機能を提供します。機械学習は、画像認識や自然言語処理から、レコメンデーションシステムや不正行為検出に至るまで、さまざまな分野で幅広く応用されており、私たちの生活様式を変えつつあります。機械学習の分野にはさまざまな手法や理論があり、その中で最も影響力のある 5 つの手法は「機械学習の 5 つの流派」と呼ばれています。 5 つの主要な学派は、象徴学派、コネクショニスト学派、進化学派、ベイジアン学派、およびアナロジー学派です。 1. 象徴主義は、象徴主義とも呼ばれ、論理的推論と知識の表現のためのシンボルの使用を強調します。この学派は、学習は既存の既存の要素を介した逆演繹のプロセスであると信じています。
 OpenAI データは必要ありません。大規模なコード モデルのリストに加わりましょう。 UIUC が StarCoder-15B-Instruct をリリース
Jun 13, 2024 pm 01:59 PM
OpenAI データは必要ありません。大規模なコード モデルのリストに加わりましょう。 UIUC が StarCoder-15B-Instruct をリリース
Jun 13, 2024 pm 01:59 PM
ソフトウェア テクノロジの最前線に立つ UIUC Zhang Lingming のグループは、BigCode 組織の研究者とともに、最近 StarCoder2-15B-Instruct 大規模コード モデルを発表しました。この革新的な成果により、コード生成タスクにおいて大きな進歩が達成され、CodeLlama-70B-Instruct を上回り、コード生成パフォーマンス リストのトップに到達しました。 StarCoder2-15B-Instruct のユニークな特徴は、その純粋な自己調整戦略であり、トレーニング プロセス全体がオープンで透過的で、完全に自律的で制御可能です。このモデルは、高価な手動アノテーションに頼ることなく、StarCoder-15B 基本モデルの微調整に応じて、StarCoder2-15B を介して数千の命令を生成します。
 フラッシュ アテンションは安定していますか?メタとハーバードは、モデルの重みの偏差が桁違いに変動していることを発見しました
May 30, 2024 pm 01:24 PM
フラッシュ アテンションは安定していますか?メタとハーバードは、モデルの重みの偏差が桁違いに変動していることを発見しました
May 30, 2024 pm 01:24 PM
MetaFAIR はハーバード大学と協力して、大規模な機械学習の実行時に生成されるデータの偏りを最適化するための新しい研究フレームワークを提供しました。大規模な言語モデルのトレーニングには数か月かかることが多く、数百、さらには数千の GPU を使用することが知られています。 LLaMA270B モデルを例にとると、そのトレーニングには合計 1,720,320 GPU 時間が必要です。大規模なモデルのトレーニングには、これらのワークロードの規模と複雑さにより、特有のシステム上の課題が生じます。最近、多くの機関が、SOTA 生成 AI モデルをトレーニングする際のトレーニング プロセスの不安定性を報告しています。これらは通常、損失スパイクの形で現れます。たとえば、Google の PaLM モデルでは、トレーニング プロセス中に最大 20 回の損失スパイクが発生しました。数値的なバイアスがこのトレーニングの不正確さの根本原因です。
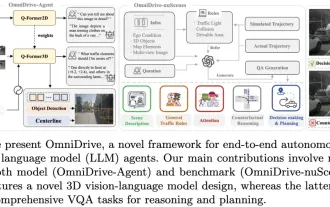 LLMはすべて完了しました! OmniDrive: 3D 認識と推論プランニングの統合 (NVIDIA の最新)
May 09, 2024 pm 04:55 PM
LLMはすべて完了しました! OmniDrive: 3D 認識と推論プランニングの統合 (NVIDIA の最新)
May 09, 2024 pm 04:55 PM
上記と著者の個人的な理解: この論文は、自動運転アプリケーションにおける現在のマルチモーダル大規模言語モデル (MLLM) の主要な課題、つまり MLLM を 2D 理解から 3D 空間に拡張する問題の解決に特化しています。自動運転車 (AV) は 3D 環境について正確な決定を下す必要があるため、この拡張は特に重要です。 3D 空間の理解は、情報に基づいて意思決定を行い、将来の状態を予測し、環境と安全に対話する車両の能力に直接影響を与えるため、AV にとって重要です。現在のマルチモーダル大規模言語モデル (LLaVA-1.5 など) は、ビジュアル エンコーダーの解像度制限や LLM シーケンス長の制限により、低解像度の画像入力しか処理できないことがよくあります。ただし、自動運転アプリケーションには次の要件が必要です。
