

AI創薬研究者がNatureサブジャーナルに参加: 専門知識を活用して創薬を加速

創薬は、化学と生物学の多くの下位分野が交差する複雑な複数段階のプロセスです。ヒト医薬品化学者は、長年にわたって蓄積した専門知識を活かして、このプロセスにおいて重要な役割を果たしています。
それでは、人工知能 (AI) は創薬において医薬品化学者が果たす役割を果たすことができるのでしょうか?答えは「はい」かもしれません。
最近、ノバルティス生物医学研究所 (NIBR) とマイクロソフト科学知能研究センター (AI4Science) の研究チームは、プロの化学者の研究を部分的に再現できる機械学習モデルを共同で提案しました。科学者の間に蓄積された知識は、しばしば「化学的直観」と呼ばれます。
研究チームは、この手法を分子モデリングの補完として使用して、将来の医薬品開発の効率を向上させることができると考えています研究論文のタイトルは「嗜好機械学習による医薬品化学の直観の抽出」で、Nature のサブジャーナルである Nature Communications に掲載されました。

医薬化学者は、ウェットラボとコンピューターの両方で、どの化合物を合成し、その後最適化する必要があるかを判断するよう求められることが多いため、創薬の「リードの最適化」段階で重要な役割を果たしています。評価は回診中に行われます。
これを行うために、医薬品化学者は通常、活性、ADMET2、または標的構造情報などの化合物の特性を含むデータを検討します。したがって、プロジェクトの成功は、生成される実験データの品質だけでなく、医薬化学のチームによる意思決定の堅牢性と合理性にも依存します。
医薬品化学者は、初期段階の創薬のさまざまな反復で何が成功するかを直感的に理解するために専門知識を活用することが多いため、より効率的に意思決定を行うことができます。これまでにも、ルールベースのアプローチや単純な化学情報学的実行可能性スコアを使用してこの知識を形式化する試みが行われてきましたが、医薬品化学者によるスコアリングに伴う微妙な点や複雑さを把握することは依然として根本的な課題です
この目標を達成するために、この研究は専門家の知識を機械学習モデルの一部に変換することを目的としています。このモデルは、リードの最適化や創薬のその他の側面における意思決定プロセスの導入において、業界で報告されている他の推奨システムと同様に補助ツールとして使用できます。
現在の医薬化学は主に手作業に依存していることを考えると、主観的なバイアスの影響を受けることは避けられません。いくつかの研究では、医薬品化学者間だけでなく、医薬品化学者内でも評価の一致度が低いと報告されています。この研究では、研究者らはマルチプレイヤー ゲームから戦略を借用することでいくつかの問題を解決したいと考えています。
彼らは、一連の分子をランク付けするタスクを好みの学習問題として捉え、シンプルなニューラル ネットワークを使用して個人の好みをモデル化しました。
図 | 研究の主なアイデアの全体概略図 (出典: 論文) 具体的には、上の図に示すように、分子は競争ゲームの参加者とみなされ、一方が勝つ確率は化学者によって提供されるフィードバックによって決定されます。これを行うために、医薬品化学者は、Web アプリケーション上で事前に指定された質問プロンプトに答え、2 つの分子のうち 1 つを選択します。合計 35 人のノバルティス医薬化学者がこのプロセスに関与し、合計 5,000 を超える注釈が付けられました。 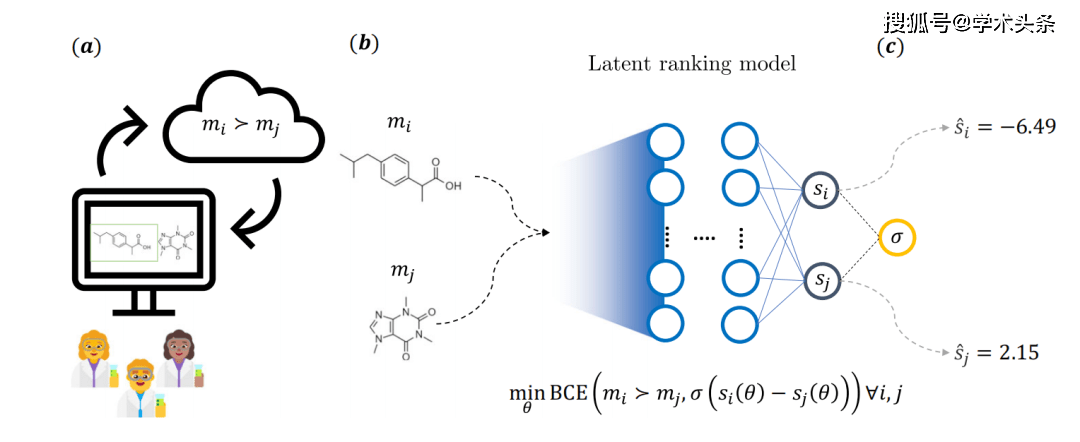 このフィードバックにより、2 つの独立したニューラル ネットワーク構造を持つモデルを使用する暗黙的なスコアリング モデルが生まれました。各分岐には固定の重みがあり、分子は一般的なケモインフォマティクス記述子を使用して特徴付けられます。トレーニング中に、モデルのパラメーターはバイナリ クロスエントロピー損失 (BCE 損失) を介して最適化されます。これは、分子ペアの基礎となるスコア差と化学者によって提供されるフィードバックに依存します。
このフィードバックにより、2 つの独立したニューラル ネットワーク構造を持つモデルを使用する暗黙的なスコアリング モデルが生まれました。各分岐には固定の重みがあり、分子は一般的なケモインフォマティクス記述子を使用して特徴付けられます。トレーニング中に、モデルのパラメーターはバイナリ クロスエントロピー損失 (BCE 損失) を介して最適化されます。これは、分子ペアの基礎となるスコア差と化学者によって提供されるフィードバックに依存します。
さらに、このモデルは、異なる薬剤間の類似性をより正確に判断できます。この研究で提案された学習スコアリング関数は、従来の薬物類似性評価指数 (QED)
よりも正確です。
研究の再現性とこの分野のさらなる発展を促進するために、研究者は「MolSkill」と呼ばれるソフトウェア パッケージも提供していることは注目に値します。これにはモデルと匿名化応答データが含まれています。
医化学分野における機械学習の問題点と応用ただし、このモデルは医薬品化学者が仕事で蓄積した知識を再現できますが、いくつかの制限もあります。 まず、化学的な直観を捉えるために、データ収集中に尋ねられる質問は常に漠然としていました。 また、提案された研究デザインでは、以前の研究と比較して参加者間の一致が大きくなりましたが、一対比較方法は完全ではありません。 さらに、「フラットランドの誤謬」は、高次元の問題を認知的に追跡できる小さな変数のセットに単純化する人間の傾向につながり、この単純化は各医薬品化学者の個人的な特性によって影響を受ける可能性があります ただし、研究チームは、この研究で提案されたモデルは現在の研究の適用範囲に限定されないと述べています。 具体的には、ここで説明したフレームワークは、創薬分野における他の定量化可能だが高価な観察対象にも拡張できます。さらに、化学分野のまだ未踏の領域についての洞察を提供できます。 これを考慮すると、研究チームは、一部の一般的なルールベースのフィルターは、人工的に生成されたトレーニング データから学習して、同様のアーキテクチャを構築できると考えています。このモデルは、推論を行う前に化合物を手動でフィルタリングする必要があるという大きな制限を克服できます。
もう 1 つ再表現する必要があるのは、特定のターゲットに対する前向きの一次最適化シナリオでは、妥当性をテストするために複数の情報源 (生物学的特性、ADMET など) を包括的に考慮する必要があるということです。研究フレームワークの実用性
 研究チームは論文の中で次のように書いています:「機械学習手法は何千もの化合物を設計でき、ハイスループットスクリーニングなどの技術は創薬プロセスの初期段階で多数の候補化合物を明らかにすることができます。提案されたスコアは「今回のメソッドは、手動検査を必要とせずに化合物をスクリーニングするために化学者の直感を暗黙的に組み込むために使用されています。このアプリケーションにより、今後数年間でメソッドの採用と信頼が加速されることが期待されています。」
研究チームは論文の中で次のように書いています:「機械学習手法は何千もの化合物を設計でき、ハイスループットスクリーニングなどの技術は創薬プロセスの初期段階で多数の候補化合物を明らかにすることができます。提案されたスコアは「今回のメソッドは、手動検査を必要とせずに化合物をスクリーニングするために化学者の直感を暗黙的に組み込むために使用されています。このアプリケーションにより、今後数年間でメソッドの採用と信頼が加速されることが期待されています。」
以上がAI創薬研究者がNatureサブジャーナルに参加: 専門知識を活用して創薬を加速の詳細内容です。詳細については、PHP 中国語 Web サイトの他の関連記事を参照してください。

ホットAIツール

Undresser.AI Undress
リアルなヌード写真を作成する AI 搭載アプリ

AI Clothes Remover
写真から衣服を削除するオンライン AI ツール。

Undress AI Tool
脱衣画像を無料で

Clothoff.io
AI衣類リムーバー

AI Hentai Generator
AIヘンタイを無料で生成します。

人気の記事
ホットツール

メモ帳++7.3.1
使いやすく無料のコードエディター

SublimeText3 中国語版
中国語版、とても使いやすい

ゼンドスタジオ 13.0.1
強力な PHP 統合開発環境

ドリームウィーバー CS6
ビジュアル Web 開発ツール

SublimeText3 Mac版
神レベルのコード編集ソフト(SublimeText3)
ホットトピック
 7532
7532
 15
1379
52
82
11
21
82
15
1379
52
82
11
21
82

 カーソルAIでバイブコーディングを試してみましたが、驚くべきことです!
Mar 20, 2025 pm 03:34 PM
カーソルAIでバイブコーディングを試してみましたが、驚くべきことです!
Mar 20, 2025 pm 03:34 PM
バイブコーディングは、無限のコード行の代わりに自然言語を使用してアプリケーションを作成できるようにすることにより、ソフトウェア開発の世界を再構築しています。 Andrej Karpathyのような先見の明に触発されて、この革新的なアプローチは開発を許可します
 2025年2月のトップ5 Genai発売:GPT-4.5、Grok-3など!
Mar 22, 2025 am 10:58 AM
2025年2月のトップ5 Genai発売:GPT-4.5、Grok-3など!
Mar 22, 2025 am 10:58 AM
2025年2月は、生成AIにとってさらにゲームを変える月であり、最も期待されるモデルのアップグレードと画期的な新機能のいくつかをもたらしました。 Xai’s Grok 3とAnthropic's Claude 3.7 SonnetからOpenaiのGまで
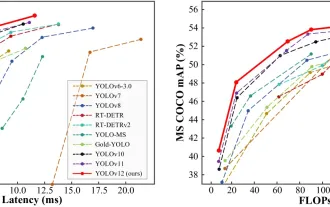 オブジェクト検出にYolo V12を使用する方法は?
Mar 22, 2025 am 11:07 AM
オブジェクト検出にYolo V12を使用する方法は?
Mar 22, 2025 am 11:07 AM
Yolo(あなたは一度だけ見ています)は、前のバージョンで各反復が改善され、主要なリアルタイムオブジェクト検出フレームワークでした。最新バージョンYolo V12は、精度を大幅に向上させる進歩を紹介します
 クリエイティブプロジェクトのための最高のAIアートジェネレーター(無料&有料)
Apr 02, 2025 pm 06:10 PM
クリエイティブプロジェクトのための最高のAIアートジェネレーター(無料&有料)
Apr 02, 2025 pm 06:10 PM
この記事では、トップAIアートジェネレーターをレビューし、その機能、創造的なプロジェクトへの適合性、価値について説明します。 Midjourneyを専門家にとって最高の価値として強調し、高品質でカスタマイズ可能なアートにDall-E 2を推奨しています。
 ChatGpt 4 oは利用できますか?
Mar 28, 2025 pm 05:29 PM
ChatGpt 4 oは利用できますか?
Mar 28, 2025 pm 05:29 PM
CHATGPT 4は現在利用可能で広く使用されており、CHATGPT 3.5のような前任者と比較して、コンテキストを理解し、一貫した応答を生成することに大幅な改善を示しています。将来の開発には、よりパーソナライズされたインターが含まれる場合があります
 chatgptよりも優れたAIはどれですか?
Mar 18, 2025 pm 06:05 PM
chatgptよりも優れたAIはどれですか?
Mar 18, 2025 pm 06:05 PM
この記事では、Lamda、Llama、GrokのようなChatGptを超えるAIモデルについて説明し、正確性、理解、業界への影響における利点を強調しています(159文字)
 次のラグモデルにミストラルOCRを使用する方法
Mar 21, 2025 am 11:11 AM
次のラグモデルにミストラルOCRを使用する方法
Mar 21, 2025 am 11:11 AM
Mistral OCR:マルチモーダルドキュメントの理解により、検索された世代の革命を起こします 検索された生成(RAG)システムはAI機能を大幅に進めており、より多くの情報に基づいた応答のために膨大なデータストアにアクセスできるようになりました
 トップAIライティングアシスタントは、コンテンツの作成を後押しします
Apr 02, 2025 pm 06:11 PM
トップAIライティングアシスタントは、コンテンツの作成を後押しします
Apr 02, 2025 pm 06:11 PM
この記事では、Grammarly、Jasper、Copy.ai、Writesonic、RytrなどのトップAIライティングアシスタントについて説明し、コンテンツ作成のためのユニークな機能に焦点を当てています。 JasperがSEOの最適化に優れているのに対し、AIツールはトーンの維持に役立つと主張します
