

北京大学と Wangshi Intelligence は、化学反応の事前トレーニングと条件付き分子生成の間のギャップを埋める新しいモデルを提案します。

化学反応は、創薬および有機化学研究の基礎です。研究コミュニティの間では、化学反応の基本的な規則を効果的に捉えることができる大規模な深層学習フレームワークに対するニーズが高まっています。
最近、北京大学と Wangshi Intelligence の研究チームは、反応ベースの分子事前トレーニングと生成タスクの間のギャップを埋める新しい方法を提案しました。
研究者たちは、有機化学のメカニズムに触発されて、モデルに帰納的バイアスを組み込むことを可能にする新しい事前トレーニング フレームワークを開発しました。この提案されたフレームワークは、困難な下流タスクを実行する際に最先端の結果を達成します。このフレームワークは、化学の知識を活用することで、少数の反応テンプレートに依存する現在の分子生成モデルの制限を克服します。広範な実験を通じて、モデルは高品質で合成可能な薬物のような構造を生成しました
全体として、この研究はさまざまな反応ベースのアプリケーションのための大規模な深層学習につながります。重要な一歩を踏み出しました。
研究のタイトルは「統合モデルによる化学反応の事前トレーニングと条件付き分子生成の間のギャップの橋渡し」で、2023 年 12 月 5 日に「Nature Machine Intelligence」に掲載されました。

#論文リンク: https://www.nature.com/articles/s42256-023-00764-9
ディープ ラーニング モデルは、多くの科学研究分野で広く使用されています。事前トレーニング フレームワークは、新しいタスクのシームレスな統合において積極的な役割を果たし、特にラベル付きデータが限られている場合にモデリング プロセスをスピードアップできます
創薬と有機化学の基礎研究 それは化学反応です。現在、データマイニングの研究と応用により、深層学習モデルを化学反応に使用できるようになりました。これらのデータに基づいて、化学反応の表現学習を詳しく調査するデータ駆動型の研究が数多く行われてきました。
表現学習とは、データから有用な特徴を自動的に学習し、それをさまざまなダウンストリームに使用することを指します。タスク。既存の方法は有機化学の基本理論を無視しており、その性能が制限されています。
化学反応に基づく分子生成反応分類タスクに加えて、化学反応に基づく分子生成も重要なアプリケーションです。以前の研究では、テンプレートベースの段階的な分子生成戦略がよく採用されていました。
これらのテンプレートベースの方法は、事前に定義された構成要素と反応に大きく依存しており、アクセス可能な化学空間が狭まっています。同様の傾向は反応生成物予測の分野でも見られ、テンプレートベースの方法では複雑な反応を外挿できませんが、この問題はテンプレートフリーの方法を使用することで解決できます。
反応ベースの分子生成タスクでは、テンプレートを使用しないメソッドは、テンプレート ベースのメソッドよりも一般化の利点も示します。ただし、既存のテンプレートフリー分子生成方法では、事前に定義された反応物ライブラリーに基づいて分子しか生成できません。これに加えて、医薬品設計におけるリード化合物またはリード最適化段階では、特定の構造を変更するための編集ツールとして化学反応を利用することがより有利です。結果として得られる化学ライブラリは、より少ない反応ステップで合成できる化学空間のサブセットに焦点を当てています。 #化学反応のための新しい包括的な深層学習フレームワーク
ここで、研究者は Uni -RXN と呼ばれる化学反応のための新しい包括的な深層学習フレームワークを提案します。これは、自己教師あり表現学習と条件付き生成モデリングという 2 つの基本タスクを解決することを目的としています。
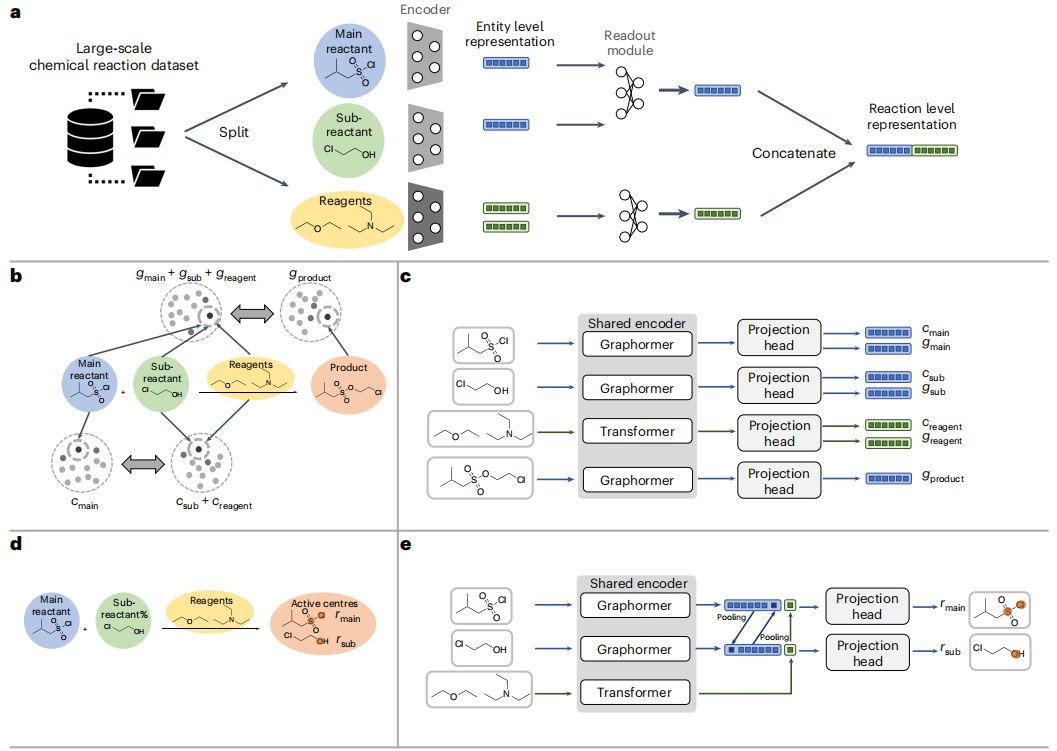 # 表示: Uni-RXN の構成と方法。 (引用元: 論文)
# 表示: Uni-RXN の構成と方法。 (引用元: 論文)
既存の方法とは異なり、研究者らは化学反応専用に設計された一連の自己教師ありタスクを提案しました。これらのタスクには、反応中心の予測、一次反応物と副反応物のペアリング、反応物と生成物のペアリングが含まれます。困難な反応タスクに関する広範な評価において、Uni-RXN メソッドは最先端技術を上回り、化学反応のドメイン知識を効果的に取得できる能力を実証しました。得られた有望な結果は、広範囲にわたる下流アプリケーションへの道を切り開きます
Uni-RXN は、化学ルールを効果的に捕捉することにより、生成タスクに最適です。事前に定義された反応物のライブラリからフラグメントを選択することに依存する従来の方法とは異なり、Uni-RXN は分子構造を入力条件として受け取り、反応内の順列不変性を維持しながら、対応する反応物の表現を生成します。 Uni-RXN は、高密度ベクトル類似性検索パッケージの機能を活用して、大規模な反応物および試薬ライブラリから反応物を効率的に検索できます。その後、反応予測モデルを使用して製品出力が生成されます。
化学空間の限られたサブセットのみを探索するテンプレートベースの方法と比較して、Uni-RXN は、より広範囲の合成可能な薬物様構造の生成において優れたパフォーマンスを示します。この機能は、仮想ライブラリの列挙に特に適しており、包括的な統計分析とケーススタディによってサポートされています。
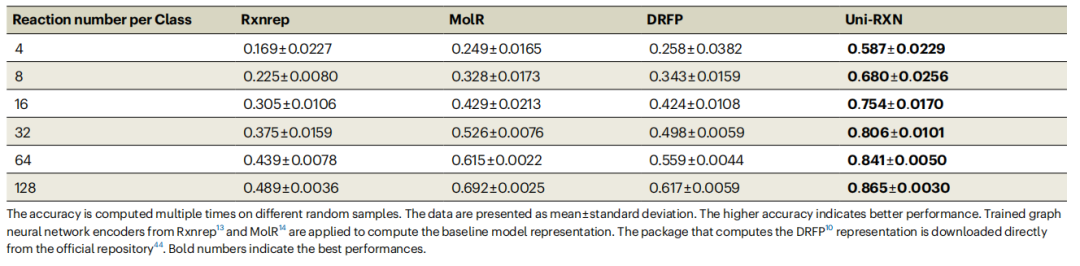
Uni-RXN アプローチには多くの利点があり、困難な化学反応分類タスクに対して豊富な表現を生成できます。他のベースライン モデルと比較して、Uni-RXN はカテゴリあたりわずか 4 データ ポイントで 58.7% の精度を達成します。
書き換えられた内容: 化学反応の分類精度については表 1 を参照してください。 (出典: 論文)
トランスフォーマー モデルを使用すると、最適化された化学反応データと最適化されていない化学反応データを区別できます。さらに、エンコーダは構造条件の生成にも簡単に適用できます。
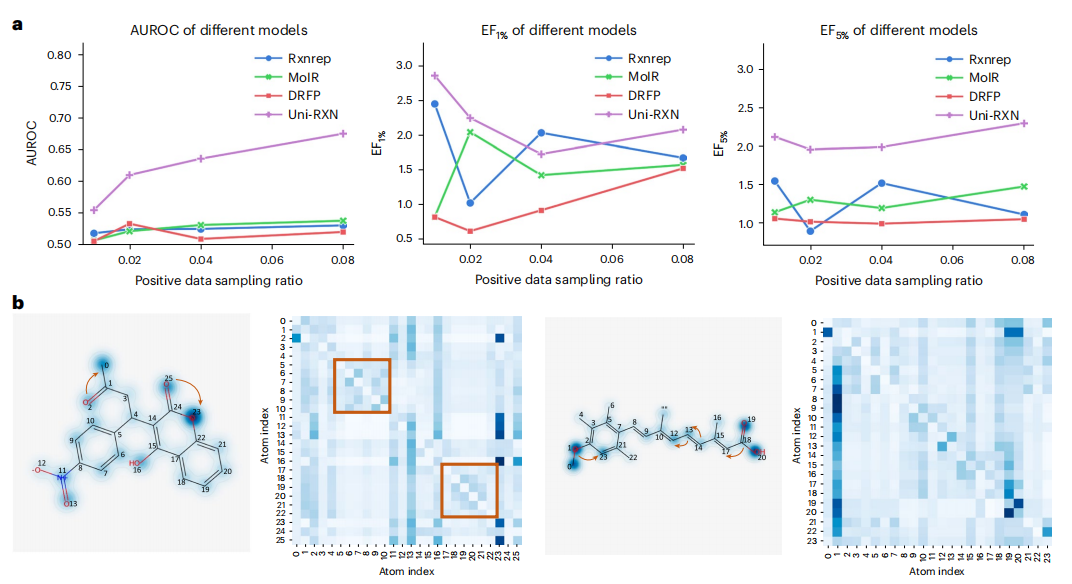
書き換える必要がある内容は次のとおりです。 Uni-RXNのリトリーブ性能と注目のウェイト。 (出典: 論文)
結果は、提案されたモデルによって生成された分子の有利な特性を強調しており、それが創薬タスクに適していることを示しています。このモデルは、薬物のような特性と合成可能性を備えたより多くの分子を生成できます。
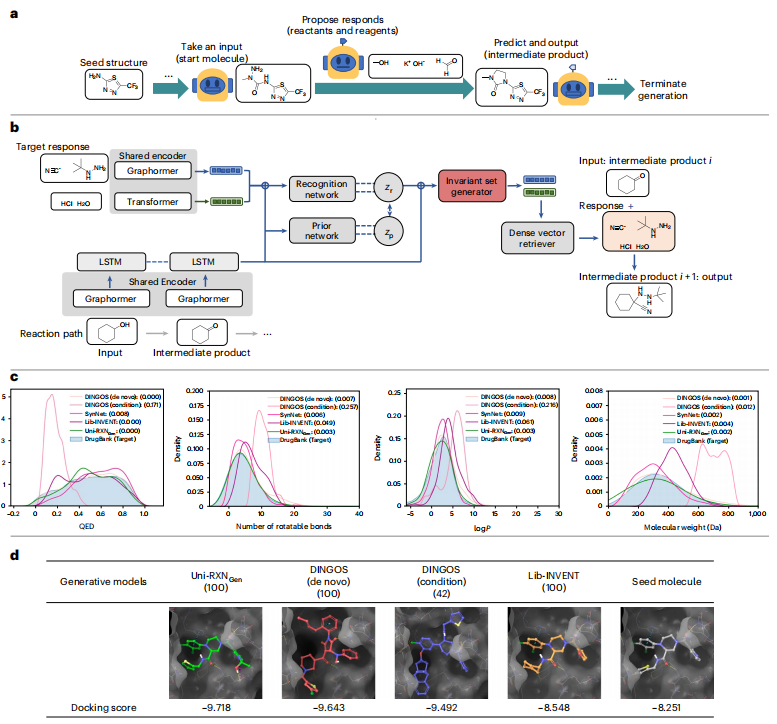
#図: Uni-RXNGen のプロセスとパフォーマンス。 (出典: 論文)
この生成されたモデルは、分子ドッキングなどの仮想スクリーニング手法と組み合わせることで、効率的な構造活性相関研究を実現できます。このモデルによって生成される巨大な合成薬物のような化学空間は、薬物の再利用またはヒット分子検索の真の陽性率を向上させることができます。
以上が北京大学と Wangshi Intelligence は、化学反応の事前トレーニングと条件付き分子生成の間のギャップを埋める新しいモデルを提案します。の詳細内容です。詳細については、PHP 中国語 Web サイトの他の関連記事を参照してください。

ホットAIツール

Undresser.AI Undress
リアルなヌード写真を作成する AI 搭載アプリ

AI Clothes Remover
写真から衣服を削除するオンライン AI ツール。

Undress AI Tool
脱衣画像を無料で

Clothoff.io
AI衣類リムーバー

AI Hentai Generator
AIヘンタイを無料で生成します。

人気の記事
ホットツール

メモ帳++7.3.1
使いやすく無料のコードエディター

SublimeText3 中国語版
中国語版、とても使いやすい

ゼンドスタジオ 13.0.1
強力な PHP 統合開発環境

ドリームウィーバー CS6
ビジュアル Web 開発ツール

SublimeText3 Mac版
神レベルのコード編集ソフト(SublimeText3)
ホットトピック
 7484
7484
 15
1377
52
77
11
19
38
15
1377
52
77
11
19
38

 世界で最も強力なオープンソース MoE モデルが登場。GPT-4 に匹敵する中国語機能を備え、価格は GPT-4-Turbo のわずか 1% 近くです
May 07, 2024 pm 04:13 PM
世界で最も強力なオープンソース MoE モデルが登場。GPT-4 に匹敵する中国語機能を備え、価格は GPT-4-Turbo のわずか 1% 近くです
May 07, 2024 pm 04:13 PM
従来のコンピューティングを超える能力を備えているだけでなく、より低コストでより効率的なパフォーマンスを実現する人工知能モデルを想像してみてください。これは SF ではありません。世界で最も強力なオープンソース MoE モデルである DeepSeek-V2[1] が登場しました。 DeepSeek-V2 は、経済的なトレーニングと効率的な推論の特徴を備えた強力な専門家混合 (MoE) 言語モデルです。これは 236B のパラメータで構成されており、そのうち 21B は各マーカーをアクティブにするために使用されます。 DeepSeek67B と比較して、DeepSeek-V2 はパフォーマンスが優れていると同時に、トレーニング コストを 42.5% 節約し、KV キャッシュを 93.3% 削減し、最大生成スループットを 5.76 倍に高めます。 DeepSeek は一般的な人工知能を研究する会社です
 AI が数学研究を破壊する!フィールズ賞受賞者で中国系アメリカ人の数学者が上位 11 件の論文を主導 | テレンス・タオが「いいね!」しました
Apr 09, 2024 am 11:52 AM
AI が数学研究を破壊する!フィールズ賞受賞者で中国系アメリカ人の数学者が上位 11 件の論文を主導 | テレンス・タオが「いいね!」しました
Apr 09, 2024 am 11:52 AM
AI は確かに数学を変えつつあります。最近、この問題に細心の注意を払っている陶哲軒氏が『米国数学協会会報』(米国数学協会会報)の最新号を送ってくれた。 「機械は数学を変えるのか?」というテーマを中心に、多くの数学者が意見を述べ、そのプロセス全体は火花に満ち、ハードコアで刺激的でした。著者には、フィールズ賞受賞者のアクシャイ・ベンカテシュ氏、中国の数学者鄭楽軍氏、ニューヨーク大学のコンピューター科学者アーネスト・デイビス氏、その他業界で著名な学者を含む強力な顔ぶれが揃っている。 AI の世界は劇的に変化しています。これらの記事の多くは 1 年前に投稿されたものです。
 こんにちは、電気アトラスです!ボストン・ダイナミクスのロボットが復活、180度の奇妙な動きにマスク氏も恐怖
Apr 18, 2024 pm 07:58 PM
こんにちは、電気アトラスです!ボストン・ダイナミクスのロボットが復活、180度の奇妙な動きにマスク氏も恐怖
Apr 18, 2024 pm 07:58 PM
Boston Dynamics Atlas は正式に電動ロボットの時代に突入します!昨日、油圧式アトラスが歴史の舞台から「涙ながらに」撤退したばかりですが、今日、ボストン・ダイナミクスは電動式アトラスが稼働することを発表しました。ボストン・ダイナミクス社は商用人型ロボットの分野でテスラ社と競争する決意を持っているようだ。新しいビデオが公開されてから、わずか 10 時間ですでに 100 万人以上が視聴しました。古い人が去り、新しい役割が現れるのは歴史的な必然です。今年が人型ロボットの爆発的な年であることは間違いありません。ネットユーザーは「ロボットの進歩により、今年の開会式は人間のように見え、人間よりもはるかに自由度が高い。しかし、これは本当にホラー映画ではないのか?」とコメントした。ビデオの冒頭では、アトラスは仰向けに見えるように地面に静かに横たわっています。次に続くのは驚くべきことです
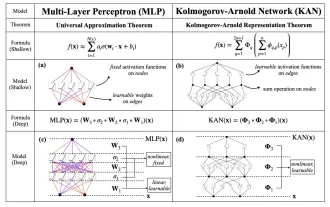 MLP に代わる KAN は、オープンソース プロジェクトによって畳み込みまで拡張されました
Jun 01, 2024 pm 10:03 PM
MLP に代わる KAN は、オープンソース プロジェクトによって畳み込みまで拡張されました
Jun 01, 2024 pm 10:03 PM
今月初め、MIT やその他の機関の研究者らは、MLP に代わる非常に有望な代替案である KAN を提案しました。 KAN は、精度と解釈可能性の点で MLP よりも優れています。また、非常に少数のパラメーターを使用して、多数のパラメーターを使用して実行する MLP よりも優れたパフォーマンスを発揮できます。たとえば、著者らは、KAN を使用して、より小規模なネットワークと高度な自動化で DeepMind の結果を再現したと述べています。具体的には、DeepMind の MLP には約 300,000 個のパラメーターがありますが、KAN には約 200 個のパラメーターしかありません。 KAN は、MLP が普遍近似定理に基づいているのに対し、KAN はコルモゴロフ-アーノルド表現定理に基づいているのと同様に、強力な数学的基礎を持っています。以下の図に示すように、KAN は
 Google は大喜び: JAX のパフォーマンスが Pytorch や TensorFlow を上回りました! GPU 推論トレーニングの最速の選択肢となる可能性があります
Apr 01, 2024 pm 07:46 PM
Google は大喜び: JAX のパフォーマンスが Pytorch や TensorFlow を上回りました! GPU 推論トレーニングの最速の選択肢となる可能性があります
Apr 01, 2024 pm 07:46 PM
Google が推進する JAX のパフォーマンスは、最近のベンチマーク テストで Pytorch や TensorFlow のパフォーマンスを上回り、7 つの指標で 1 位にランクされました。また、テストは最高の JAX パフォーマンスを備えた TPU では行われませんでした。ただし、開発者の間では、依然として Tensorflow よりも Pytorch の方が人気があります。しかし、将来的には、おそらくより大規模なモデルが JAX プラットフォームに基づいてトレーニングされ、実行されるようになるでしょう。モデル 最近、Keras チームは、ネイティブ PyTorch 実装を使用して 3 つのバックエンド (TensorFlow、JAX、PyTorch) をベンチマークし、TensorFlow を使用して Keras2 をベンチマークしました。まず、主流のセットを選択します
 時系列予測 NLP 大規模モデルの新機能: 時系列予測の暗黙的なプロンプトを自動的に生成
Mar 18, 2024 am 09:20 AM
時系列予測 NLP 大規模モデルの新機能: 時系列予測の暗黙的なプロンプトを自動的に生成
Mar 18, 2024 am 09:20 AM
今日は、時系列予測のパフォーマンスを向上させるために、時系列データを潜在空間上の大規模な自然言語処理 (NLP) モデルと整合させる方法を提案するコネチカット大学の最近の研究成果を紹介したいと思います。この方法の鍵は、潜在的な空間ヒント (プロンプト) を使用して時系列予測の精度を高めることです。論文タイトル: S2IP-LLM: SemanticSpaceInformedPromptLearningwithLLMforTimeSeriesForecasting ダウンロードアドレス: https://arxiv.org/pdf/2403.05798v1.pdf 1. 大きな問題の背景モデル
 テスラのロボットは工場で働く、マスク氏:手の自由度は今年22に達する!
May 06, 2024 pm 04:13 PM
テスラのロボットは工場で働く、マスク氏:手の自由度は今年22に達する!
May 06, 2024 pm 04:13 PM
テスラのロボット「オプティマス」の最新映像が公開され、すでに工場内で稼働可能となっている。通常の速度では、バッテリー(テスラの4680バッテリー)を次のように分類します:公式は、20倍の速度でどのように見えるかも公開しました - 小さな「ワークステーション」上で、ピッキング、ピッキング、ピッキング:今回は、それがリリースされたハイライトの1つビデオの内容は、オプティマスが工場内でこの作業を完全に自律的に行い、プロセス全体を通じて人間の介入なしに完了するというものです。そして、オプティマスの観点から見ると、自動エラー修正に重点を置いて、曲がったバッテリーを拾い上げたり配置したりすることもできます。オプティマスのハンドについては、NVIDIA の科学者ジム ファン氏が高く評価しました。オプティマスのハンドは、世界の 5 本指ロボットの 1 つです。最も器用。その手は触覚だけではありません
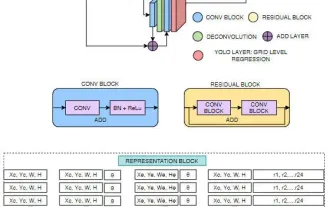 FisheyeDetNet: 魚眼カメラに基づいた最初のターゲット検出アルゴリズム
Apr 26, 2024 am 11:37 AM
FisheyeDetNet: 魚眼カメラに基づいた最初のターゲット検出アルゴリズム
Apr 26, 2024 am 11:37 AM
目標検出は自動運転システムにおいて比較的成熟した問題であり、その中でも歩行者検出は最も初期に導入されたアルゴリズムの 1 つです。ほとんどの論文では非常に包括的な研究が行われています。ただし、サラウンドビューに魚眼カメラを使用した距離認識については、あまり研究されていません。放射状の歪みが大きいため、標準のバウンディング ボックス表現を魚眼カメラに実装するのは困難です。上記の説明を軽減するために、拡張バウンディング ボックス、楕円、および一般的な多角形の設計を極/角度表現に探索し、これらの表現を分析するためのインスタンス セグメンテーション mIOU メトリックを定義します。提案された多角形モデルの FisheyeDetNet は、他のモデルよりも優れたパフォーマンスを示し、同時に自動運転用の Valeo 魚眼カメラ データセットで 49.5% の mAP を達成しました。
