

幾何学的な深層学習手法を使用して薬物分子を合成するための最適なオプションを予測し、新薬発見への道を開く


後段階官能化は、薬剤候補の特性を最適化するための経済的な方法です。しかし、薬物分子の化学的複雑さにより、後期段階の機能化が困難になることがよくあります。
この問題を解決するために、ミュンヘン大学、チューリッヒ工科大学、ロシュ バーゼルの研究者が協力して、後期段階の機能化プラットフォームを開発しました。幾何学的深層学習とハイスループット反応スクリーニング技術に基づいています
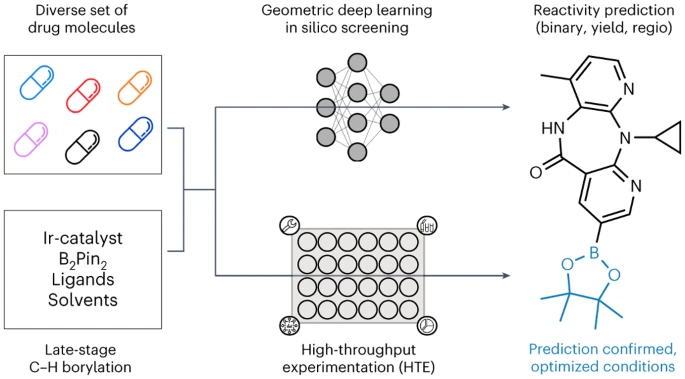
ホウ素化が機能化の重要なステップの 1 つであることを考慮して、計算モデルを使用してさまざまな反応条件下での収率を予測しました。平均絶対誤差範囲は次のとおりです。 4~5%。このモデルは、既知の基質と未知の基質に対する新しい反応を、それぞれ 92% と 67% の精度で分類できました。分類器の F スコアは 67% で、主生成物の位置選択性を正確に捕捉することができました。私たちは、23 種類の市販薬分子に適用した場合に、構造多様化の多くの機会を特定することに成功しました。
この研究のタイトルは「幾何学深層学習を使用して、後期段階の薬物多様化を促進するハイスループット実験を可能にする」であり、ジャーナルに掲載されました。 Nature Chemistry 2023 年 11 月 23 日

#LSF プロジェクトは医薬化学研究において重要な役割を果たします
構造の新規性と複雑性医薬化学における構造活性相関の確立を目指す場合、化学標的構造の合成は困難になります。構造活性相関モデルは、医薬品候補の薬理学的活性と物理化学的特性を改善するためのリード化合物とリード化合物の最適化計画を導くことができます。効率的な統合は、設計、製造、テスト、分析サイクルのボトルネックである構造活性相関の探索にとって極めて重要です。C-H 結合を活性化および修飾して有機足場を実現するための代替方法は数多くあります。後期段階機能化(LSF):分子ビルディングブロックから先進的な医薬品分子まで。多くの触媒システムは、指向性および無指向性のアプローチに加え、修飾類似体への化学的および部位選択的なアクセスを提供します。数多くの LSF 法の中で、C-H ボリル化法は、高速化合物に最も一般的に使用されると考えられています。アプローチ。有機ホウ素化合物は、その後の C-C 結合カップリング反応の信頼できる手段としてさまざまな官能基に変換できるため、広範な構造活性相関研究が可能になりますしかし、現在、創薬においては、LSF のアプリには報告が少ない。これらのレポートのほとんどは、単一の LSF 反応タイプに焦点を当てています。異なる結合強度、電子的特性、立体および官能基環境を備えた複数の種類の C-H 結合の直接 LSF には課題が生じます。さらに、LSF プロジェクトの実施は時間とリソースを大量に消費することが多く、多くの医薬化学プロジェクトのタイトなスケジュールと限られた資産とは一致しません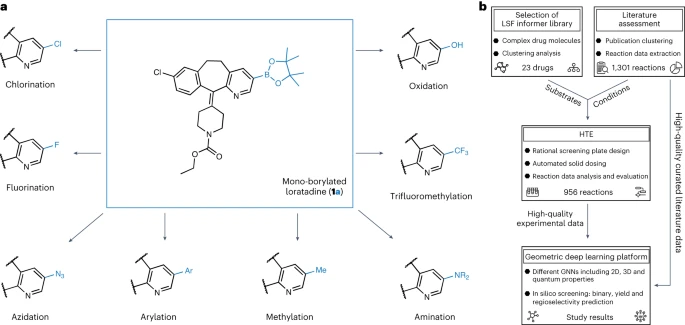
人工知能対応LSF(言語サポート機能)
ハイスループット実験(HTE)は確立された反応最適化手法です。 、小型化された低バッチスクリーニングが可能であり、少数の貴重な構成要素と消耗品を使用して複数の変換を迅速かつ再現性よく並行して実行できます。 HTE は、応答の成功と失敗に関する高品質のデータセットを生成する FAIR (検索可能性、アクセシビリティ、相互運用性、再利用性) ドキュメントと組み合わせることで、高度なデータ分析と機械学習を可能にし、創薬のための LSF を可能にします。 グラフ ニューラル ネットワーク (GNN) は、分子の特徴抽出と属性予測に広く使用されています。化学反応計画のために開発されたさまざまな機械学習手法の中で、GNN は逆合成計画、位置選択性予測、反応生成物の予測に適用されて成功しています。さらに、同様の問題を解決するために、トランスフォーマーやフィンガープリントに基づく方法も開発されています。研究によると、遷移状態の幾何学構造を学習することで、競合する反応の結果を正確に予測できることが示されています。電子効果によって駆動される反応の位置選択性の予測は、密度汎関数理論 (DFT) と原子の部分電荷のグラフによる特性評価を使用して改善できます。グラフ機械学習とハイスループット実験 (HTE) を組み合わせることで、有機基板の C-H 活性化反応の条件を最適化できます。一部の研究では、場合によってはエナンチオ選択性を含む反応結果を予測する能力を持つ遷移状態の深層学習モデルの使用に焦点を当てていますが、これらの方法は小分子構造および比較的小さな分子に限定されています。このようなモデルをより構造的に複雑な薬物様分子に適用することは困難です。文献研究に基づいて、イリジウム触媒によるホウ素化反応の位置選択性は、遷移状態の量子化学情報で強化されたハイブリッド機械学習モデルを通じて予測できます。ただし、C-H 活性化反応モデルのパフォーマンスと複数の芳香環系を持つ分子における位置選択的応用に対する立体効果と電子効果の影響はまだ調査されていません。幾何学深層学習による自動 LSF ホウ素スクリーニング
ミュンヘン大学、チューリッヒ工科大学、ロシュ ファーマシューティカルズ バーゼルの研究者は、幾何学深層学習 LSF ホウ素に適用された自動 LSF ホウ素スクリーニング手法を発表します。後期段階のヒット商品とリード多様化の機会を特定するためのスクリーニング方法。コンピューターによる深層学習を利用して、複雑な薬物分子 LSF の反応結果、収率、位置選択性を予測しました。
以上が幾何学的な深層学習手法を使用して薬物分子を合成するための最適なオプションを予測し、新薬発見への道を開くの詳細内容です。詳細については、PHP 中国語 Web サイトの他の関連記事を参照してください。

ホットAIツール

Undresser.AI Undress
リアルなヌード写真を作成する AI 搭載アプリ

AI Clothes Remover
写真から衣服を削除するオンライン AI ツール。

Undress AI Tool
脱衣画像を無料で

Clothoff.io
AI衣類リムーバー

AI Hentai Generator
AIヘンタイを無料で生成します。

人気の記事
ホットツール

メモ帳++7.3.1
使いやすく無料のコードエディター

SublimeText3 中国語版
中国語版、とても使いやすい

ゼンドスタジオ 13.0.1
強力な PHP 統合開発環境

ドリームウィーバー CS6
ビジュアル Web 開発ツール

SublimeText3 Mac版
神レベルのコード編集ソフト(SublimeText3)
ホットトピック
 7510
7510
 15
1378
52
78
11
19
64
15
1378
52
78
11
19
64

 DeepMind ロボットが卓球をすると、フォアハンドとバックハンドが空中に滑り出し、人間の初心者を完全に打ち負かしました
Aug 09, 2024 pm 04:01 PM
DeepMind ロボットが卓球をすると、フォアハンドとバックハンドが空中に滑り出し、人間の初心者を完全に打ち負かしました
Aug 09, 2024 pm 04:01 PM
でももしかしたら公園の老人には勝てないかもしれない?パリオリンピックの真っ最中で、卓球が注目を集めています。同時に、ロボットは卓球のプレーにも新たな進歩をもたらしました。先ほど、DeepMind は、卓球競技において人間のアマチュア選手のレベルに到達できる初の学習ロボット エージェントを提案しました。論文のアドレス: https://arxiv.org/pdf/2408.03906 DeepMind ロボットは卓球でどれくらい優れていますか?おそらく人間のアマチュアプレーヤーと同等です: フォアハンドとバックハンドの両方: 相手はさまざまなプレースタイルを使用しますが、ロボットもそれに耐えることができます: さまざまなスピンでサーブを受ける: ただし、ゲームの激しさはそれほど激しくないようです公園の老人。ロボット、卓球用
 初のメカニカルクロー!元羅宝は2024年の世界ロボット会議に登場し、家庭に入ることができる初のチェスロボットを発表した
Aug 21, 2024 pm 07:33 PM
初のメカニカルクロー!元羅宝は2024年の世界ロボット会議に登場し、家庭に入ることができる初のチェスロボットを発表した
Aug 21, 2024 pm 07:33 PM
8月21日、2024年世界ロボット会議が北京で盛大に開催された。 SenseTimeのホームロボットブランド「Yuanluobot SenseRobot」は、全製品ファミリーを発表し、最近、世界初の家庭用チェスロボットとなるYuanluobot AIチェスプレイロボット - Chess Professional Edition(以下、「Yuanluobot SenseRobot」という)をリリースした。家。 Yuanluobo の 3 番目のチェス対局ロボット製品である新しい Guxiang ロボットは、AI およびエンジニアリング機械において多くの特別な技術アップグレードと革新を経て、初めて 3 次元のチェスの駒を拾う機能を実現しました。家庭用ロボットの機械的な爪を通して、チェスの対局、全員でのチェスの対局、記譜のレビューなどの人間と機械の機能を実行します。
 クロードも怠け者になってしまった!ネチズン: 自分に休日を与える方法を学びましょう
Sep 02, 2024 pm 01:56 PM
クロードも怠け者になってしまった!ネチズン: 自分に休日を与える方法を学びましょう
Sep 02, 2024 pm 01:56 PM
もうすぐ学校が始まり、新学期を迎える生徒だけでなく、大型AIモデルも気を付けなければなりません。少し前、レディットはクロードが怠け者になったと不満を漏らすネチズンでいっぱいだった。 「レベルが大幅に低下し、頻繁に停止し、出力も非常に短くなりました。リリースの最初の週は、4 ページの文書全体を一度に翻訳できましたが、今では 0.5 ページの出力さえできません」 !」 https://www.reddit.com/r/ClaudeAI/comments/1by8rw8/something_just_feels_wrong_with_claude_in_the/ というタイトルの投稿で、「クロードには完全に失望しました」という内容でいっぱいだった。
 世界ロボット会議で「未来の高齢者介護の希望」を担う家庭用ロボットを囲みました
Aug 22, 2024 pm 10:35 PM
世界ロボット会議で「未来の高齢者介護の希望」を担う家庭用ロボットを囲みました
Aug 22, 2024 pm 10:35 PM
北京で開催中の世界ロボット会議では、人型ロボットの展示が絶対的な注目となっているスターダストインテリジェントのブースでは、AIロボットアシスタントS1がダルシマー、武道、書道の3大パフォーマンスを披露した。文武両道を備えた 1 つの展示エリアには、多くの専門的な聴衆とメディアが集まりました。弾性ストリングのエレガントな演奏により、S1 は、スピード、強さ、正確さを備えた繊細な操作と絶対的なコントロールを発揮します。 CCTVニュースは、「書道」の背後にある模倣学習とインテリジェント制御に関する特別レポートを実施し、同社の創設者ライ・ジエ氏は、滑らかな動きの背後にあるハードウェア側が最高の力制御と最も人間らしい身体指標(速度、負荷)を追求していると説明した。など)、AI側では人の実際の動きのデータが収集され、強い状況に遭遇したときにロボットがより強くなり、急速に進化することを学習することができます。そしてアジャイル
 ACL 2024 賞の発表: HuaTech による Oracle 解読に関する最優秀論文の 1 つ、GloVe Time Test Award
Aug 15, 2024 pm 04:37 PM
ACL 2024 賞の発表: HuaTech による Oracle 解読に関する最優秀論文の 1 つ、GloVe Time Test Award
Aug 15, 2024 pm 04:37 PM
貢献者はこの ACL カンファレンスから多くのことを学びました。 6日間のACL2024がタイのバンコクで開催されています。 ACL は、計算言語学と自然言語処理の分野におけるトップの国際会議で、国際計算言語学協会が主催し、毎年開催されます。 ACL は NLP 分野における学術的影響力において常に第一位にランクされており、CCF-A 推奨会議でもあります。今年の ACL カンファレンスは 62 回目であり、NLP 分野における 400 以上の最先端の作品が寄せられました。昨日の午後、カンファレンスは最優秀論文およびその他の賞を発表しました。今回の優秀論文賞は7件(未発表2件)、最優秀テーマ論文賞1件、優秀論文賞35件です。このカンファレンスでは、3 つの Resource Paper Award (ResourceAward) と Social Impact Award (
 宏蒙スマートトラベルS9とフルシナリオ新製品発売カンファレンス、多数の大ヒット新製品が一緒にリリースされました
Aug 08, 2024 am 07:02 AM
宏蒙スマートトラベルS9とフルシナリオ新製品発売カンファレンス、多数の大ヒット新製品が一緒にリリースされました
Aug 08, 2024 am 07:02 AM
今日の午後、Hongmeng Zhixingは新しいブランドと新車を正式に歓迎しました。 8月6日、ファーウェイはHongmeng Smart Xingxing S9およびファーウェイのフルシナリオ新製品発表カンファレンスを開催し、パノラマスマートフラッグシップセダンXiangjie S9、新しいM7ProおよびHuawei novaFlip、MatePad Pro 12.2インチ、新しいMatePad Air、Huawei Bisheng Withを発表しました。レーザー プリンタ X1 シリーズ、FreeBuds6i、WATCHFIT3、スマート スクリーン S5Pro など、スマート トラベル、スマート オフィスからスマート ウェアに至るまで、多くの新しいオールシナリオ スマート製品を開発し、ファーウェイは消費者にスマートな体験を提供するフル シナリオのスマート エコシステムを構築し続けています。すべてのインターネット。宏孟志興氏:スマートカー業界のアップグレードを促進するための徹底的な権限付与 ファーウェイは中国の自動車業界パートナーと提携して、
 Li Feifei 氏のチームは、ロボットに空間知能を与え、GPT-4o を統合する ReKep を提案しました
Sep 03, 2024 pm 05:18 PM
Li Feifei 氏のチームは、ロボットに空間知能を与え、GPT-4o を統合する ReKep を提案しました
Sep 03, 2024 pm 05:18 PM
ビジョンとロボット学習の緊密な統合。最近話題の1X人型ロボットNEOと合わせて、2つのロボットハンドがスムーズに連携して服をたたむ、お茶を入れる、靴を詰めるといった動作をしていると、いよいよロボットの時代が到来するのではないかと感じられるかもしれません。実際、これらの滑らかな動きは、高度なロボット技術 + 精緻なフレーム設計 + マルチモーダル大型モデルの成果です。有用なロボットは多くの場合、環境との複雑かつ絶妙な相互作用を必要とし、環境は空間領域および時間領域の制約として表現できることがわかっています。たとえば、ロボットにお茶を注いでもらいたい場合、ロボットはまずティーポットのハンドルを掴んで、お茶をこぼさないように垂直に保ち、次にポットの口がカップの口と揃うまでスムーズに動かす必要があります。 、そしてティーポットを一定の角度に傾けます。これ
 分散型人工知能カンファレンス DAI 2024 論文募集: エージェント デイ、強化学習の父であるリチャード サットン氏が出席します。 Yan Shuicheng、Sergey Levine、DeepMind の科学者が基調講演を行います
Aug 22, 2024 pm 08:02 PM
分散型人工知能カンファレンス DAI 2024 論文募集: エージェント デイ、強化学習の父であるリチャード サットン氏が出席します。 Yan Shuicheng、Sergey Levine、DeepMind の科学者が基調講演を行います
Aug 22, 2024 pm 08:02 PM
会議の紹介 科学技術の急速な発展に伴い、人工知能は社会の進歩を促進する重要な力となっています。この時代に、分散型人工知能 (DAI) の革新と応用を目撃し、参加できることは幸運です。分散型人工知能は人工知能分野の重要な分野であり、近年ますます注目を集めています。大規模言語モデル (LLM) に基づくエージェントは、大規模モデルの強力な言語理解機能と生成機能を組み合わせることで、自然言語対話、知識推論、タスク計画などにおいて大きな可能性を示しました。 AIAgent は大きな言語モデルを引き継ぎ、現在の AI 界隈で話題になっています。アウ
