

HMMER ソフトウェアを Windows システムにインストールできますか?


Windows システムは mmer ソフトウェアをインストールできますか
ダウンロードとインストール
Mac OS/X、Linux、UNIX システムの場合は、ソース コードからコンパイルしてインストールします:
% wget ftp://selab.janelia.org/pub/software/hmmer3/3.0/hmmer-3.0.tar.gz % tar zxf hmmer-3.0.tar.gz % cd hmmer-3.0 % ./configure % make % make check
Windows システムの場合は、バイナリ圧縮パッケージを直接ダウンロードし、解凍して使用します。
hmmer に含まれるプログラム
phmmer: Blastp と同様に、タンパク質配列を使用してタンパク質配列ライブラリを検索します;
>phmmer チュートリアル/HBB HUMAN uniprot sprot.fa
jackhmmer: psiBlast と同様に、タンパク質配列はタンパク質配列ライブラリを繰り返し検索します。
>jackhmmer チュートリアル/HBB HUMAN uniprot sprot.fahmmbuild: 複数のアライメント シーケンスを使用して HMM モデルを構築します;
hmmsearch: HMM モデルを使用して配列ライブラリを検索します;
hmmscan: シーケンスを使用して HMM ライブラリを検索します;
hmmalign: 複数のアライメント シーケンスを構築するための手がかりとして HMM を使用します;
>hmmalign globins4.hmm チュートリアル/globins45.fa
hmmconvert: HMM 形式を変換します
hmmemit: HMM モデルからパターン シーケンスを取得します;
hmmfetch: 名前または受付番号によって HMM ライブラリから HMM モデルを取得します;
hmmpress: HMM データベースをフォーマットして、hmscan の検索と使用を容易にします;
hmmstat: HMM データベースの統計情報を表示します;
HMMモデルを用いた配列データベースの検索
hmmbuild を使用して HMM モデルを構築し、ストックホルム形式または FASTA 形式のマルチプル アライメント シーケンス ファイル (tutorial/globins4.sto など) を入力します。コマンドは次のとおりです。
>hmmbuild globins4.hmm チュートリアル/globins4.sto
globins4.hmm は出力 HMM モデルです
hmmsearch を使用してタンパク質配列データベースを検索します。タンパク質配列データベースは FASTA 形式です。コマンドは次のとおりです:
>hmmsearch globins4.hmm uniprot sprot.fasta >globins4.out
globins4.out は、次のような出力結果ファイルです。
#この例では、公式チュートリアルの例を使用しています
タンパク質配列を使用して HMM データベースを検索します
HMM データベースを構築します。HMM データベースは、複数の HMM モデルを含むファイルです。Pfam、SMART、TIGRFams からダウンロードすることも、次のような複数のアライメント シーケンスから自分で構築することもできます。
>hmmbuild globins4.hmm チュートリアル/globins4.sto>hmmbuild fn3.hmm チュートリアル/fn3.sto
>hmmbuild Pkinase.hmm チュートリアル/Pkinase.sto
>cat globins4.hmm fn3.hmm Pkinase.hmm >minifam
hmmpress を使用して、圧縮やインデックスの作成を含むデータベースをフォーマットします。コマンドは次のとおりです:
>hmmpress ミニファム
このステップはすぐに完了でき、出力内容は次のとおりです。
作業中…完了しました。
3 つの HMM (3 つの名前と 2 つのアクセッション) が押されてインデックス付けされました。
モデルはバイナリ ファイルに圧縮されました: minifam.h3m
バイナリ モデル ファイルの SSI インデックス: minifam.h3i
プロファイル (MSV 部分) が圧縮されました: minifam.h3f
プロファイル (残り) が圧縮されました: minifam.h3p
hmmscan を使用して HMM データベースを検索します。コマンドは次のとおりです:
>hmmscan ミニファム チュートリアル/7LESS_DROME
hmmer ソフトウェアはどのようにして fasta 形式のファイルを sto 形式に変換しますか?
私もこの問題に遭遇しました。長時間オンラインで検索しましたが、適切な解決策が見つからなかったので、自分で解決策を書きました。コードは次のとおりです。
import glob # これらはすべて標準ライブラリからのものです
OSをインポート
# hmm をビルドしたい fasta ファイル (比較済み) をこのプログラムと同じフォルダーに置き、このプログラムを実行して hmmbuild を実行します。
os.chdir(os.path.dirname(__file__))
fs = glob.glob('*.fasta') # 各 fasta ファイルを取得します。fasta ファイルに .fasta 以外のサフィックスが付いている場合は、ここで変更するか、直接 '*.fa*' に変更できます。
fs の f の場合:
hmm = os.path.splitext(f)[0] '.hmm'
ストックホルム = os.path.splitext(f)[0] '.sto'
with open(f, 'r') as fhandle: # これは、fasta ファイルを読み取り、すべての fasta ファイルをリストに保存するために使用されます
fastas = ['>' tmp.replace('\n', '\r', 1).replace('\n', '').replace('\r', '\n') forタプルの tmp(filter(None, (fhandle.read().split('>'))))]
for i in range(len(fastas)):
fastas[i] = fastas[i].split('\n')
fastas[i][0] = fastas[i][0].split()[0][1:10]
tmp = []
for j in range(len(fastas[i][1]) // 80 1):
tmp.append(fastas[i][1][80 * j : 80 * j 80])
fastas[i][1] = tmp
with open(stockholm, 'w') as out: # sto ファイルはここに書かれています
out.write('# ストックホルム 1.0\n\n')
for j in range(len(fastas[0][1]) - 1):
for i in range(len(fastas)):
out.write('% -12s%s\n' % (fastas[i][0], fastas[i][1][j]))
out.write('\n')
for i in range(len(fastas)):
out.write('% -12s%s\n' % (fastas[i][0], fastas[i][1][-1]))
out.write('//')
os.system('hmmbuild --amino %s %s' % (hmm, Stockholm)) # hmmbuild はここで実行されており、
内のパラメータを変更できますバイオインフォマティクスを独学で学ぶ方法
1. 既存のバイオインフォマティクス ツールから始めます。生物学研究に役立つ既存のソフトウェア、ネットワーク サーバー、データベースなどの使用方法に慣れてください。作業を繰り返さないでください。既製のツールを使用できる場合は、独自のツールを開発しないでください。作ったもの。
2. DOS や Linux などのコマンド ライン オペレーティング システムに慣れていると、単純なシェルを作成でき、コマンド ライン レベルのプログラムをインストールして通常のプロセスを実行できます。ソフトウェアを見つけてインストールする方法を学ぶことは、最も重要かつ基本的なスキルです。実際、適切なソフトウェア パッケージを見つければ、多くの問題を簡単に解決できます。
3. 簡単なスクリプト言語に慣れてください。個人的には Python の使用をお勧めします。具体的な理由については、私の投稿を参照してください。小さなスクリプトは、既製のツールがない場合、またはデータ形式の変換が必要な場合に非常に役立ちます。一般的なアプリケーションは、それ自体で多くのコードを記述する必要はありませんが、私たちが通常遭遇する問題は他の専門家も遭遇している可能性があると考えなければならないため、インターネット上には多数のツールキットが存在します。その他のプログラミング言語に関しては、R や perl などはどれも似たようなもので、すべてをマスターできます。
4. 簡単なアルゴリズムやデータ構造の知識を身につけておくと、多くのプログラムの内部機構を理解し、その長所と短所を知ることができ、独自のプログラムを書く際にも役立ちます。体力があれば統計学や機械学習などを勉強しましょう。 。
5. 独自の生物学分野の範囲を拡大、研究、分析、開発します。
以上がHMMER ソフトウェアを Windows システムにインストールできますか?の詳細内容です。詳細については、PHP 中国語 Web サイトの他の関連記事を参照してください。

ホットAIツール

Undresser.AI Undress
リアルなヌード写真を作成する AI 搭載アプリ

AI Clothes Remover
写真から衣服を削除するオンライン AI ツール。

Undress AI Tool
脱衣画像を無料で

Clothoff.io
AI衣類リムーバー

AI Hentai Generator
AIヘンタイを無料で生成します。

人気の記事
ホットツール

メモ帳++7.3.1
使いやすく無料のコードエディター

SublimeText3 中国語版
中国語版、とても使いやすい

ゼンドスタジオ 13.0.1
強力な PHP 統合開発環境

ドリームウィーバー CS6
ビジュアル Web 開発ツール

SublimeText3 Mac版
神レベルのコード編集ソフト(SublimeText3)
ホットトピック
 7480
7480
 15
1377
52
77
11
19
33
15
1377
52
77
11
19
33

 Win11 Activation Key Permanent 2025
Mar 18, 2025 pm 05:57 PM
Win11 Activation Key Permanent 2025
Mar 18, 2025 pm 05:57 PM
記事では、2025年まで有効な恒久的なWindows 11キーのソース、法的問題、非公式キーを使用するリスクについて説明します。注意と合法性をアドバイスします。
 Win11 Activation Key Permanent 2024
Mar 18, 2025 pm 05:56 PM
Win11 Activation Key Permanent 2024
Mar 18, 2025 pm 05:56 PM
記事では、2024年の恒久的なWindows 11アクティベーションキーの信頼できるソース、サードパーティキーの法的意味、および非公式キーを使用するリスクについて説明します。
 Acer PD163Qデュアルポータブルモニターレビュー:私は本当にこれを愛したかった
Mar 18, 2025 am 03:04 AM
Acer PD163Qデュアルポータブルモニターレビュー:私は本当にこれを愛したかった
Mar 18, 2025 am 03:04 AM
ACER PD163Qデュアルポータブルモニター:接続の悪夢 私はAcer PD163Qに大きな期待を抱いていました。単一のケーブルを介して便利に接続するデュアルポータブルディスプレイの概念は、非常に魅力的でした。 残念ながら、この魅力的なアイデアはquicです
 トップ3のWindows11ゲームは、Windows1を覆うことを特徴としています
Mar 16, 2025 am 12:17 AM
トップ3のWindows11ゲームは、Windows1を覆うことを特徴としています
Mar 16, 2025 am 12:17 AM
Windows 11へのアップグレード:PCゲームエクスペリエンスを強化します Windows 11は、PCゲームエクスペリエンスを大幅に向上させるエキサイティングな新しいゲーム機能を提供します。 このアップグレードは、Windows 10から移動するPCゲーマーについて検討する価値があります。 Auto HDR:Eleva
 この野生のウルトラ幅のエイリアンウェアモニターは、今日300ドルです
Mar 13, 2025 pm 12:21 PM
この野生のウルトラ幅のエイリアンウェアモニターは、今日300ドルです
Mar 13, 2025 pm 12:21 PM
Alienware AW3225QF:最高の曲線4Kディスプレイ、購入する価値はありますか? Alienware AW3225QFは、最高の曲線4Kディスプレイとして知られており、その強力なパフォーマンスは疑いの余地がありません。高速応答時間、見事なHDR効果、無制限のコントラストは、優れた色のパフォーマンスと相まって、このモニターの利点です。主にゲーマーを対象としていますが、OLEDの欠点を受け入れることができれば、高効率を追求するオフィスワーカーにも適しています。 ワイドスクリーンモニターは、ゲーマーに愛されるだけでなく、生産性の向上を重視するユーザーにも好まれています。彼らは仕事に最適で、誰のデスクトップ体験を強化します。このエイリアンウェアモニターは通常高価ですが、現在それを楽しんでいます
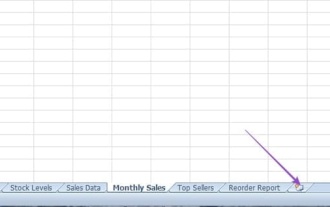 Excelで目次を作成する方法
Mar 24, 2025 am 08:01 AM
Excelで目次を作成する方法
Mar 24, 2025 am 08:01 AM
目次は、大規模なファイルを使用する際のゲームチェンジャーの合計です。これにより、すべてが整理され、ナビゲートしやすくなります。残念ながら、Wordとは異なり、Microsoft Excelにはtを追加する単純な「目次」ボタンがありません
 オープンソースのウィンドウであるReactosは、アップデートを取得しました
Mar 25, 2025 am 03:02 AM
オープンソースのウィンドウであるReactosは、アップデートを取得しました
Mar 25, 2025 am 03:02 AM
Reactos 0.4.15には、新しいストレージドライバーが含まれています。これは、全体的な安定性とUDBドライブの互換性、およびネットワークの新しいドライバーに役立つはずです。フォントサポート、デスクトップシェル、Windows API、テーマ、ファイルへの多くの更新もあります
 新しいモニターの買い物?避けるべき8つの間違い
Mar 18, 2025 am 03:01 AM
新しいモニターの買い物?避けるべき8つの間違い
Mar 18, 2025 am 03:01 AM
新しいモニターを購入することは頻繁に発生することではありません。 これは、コンピューター間を頻繁に移動する長期的な投資です。ただし、アップグレードは避けられず、最新のスクリーンテクノロジーは魅力的です。 しかし、間違った選択をすることはあなたに後悔を残すことができます
