

0보다 빠르다! Meta는 AlphaFold2를 분쇄하기 위해 150억 개의 매개변수를 갖춘 대규모 단백질 모델을 출시했습니다.

현재까지 가장 큰 단백질 언어 모델이 출시되었습니다!
1년 전 DeepMind의 오픈 소스 AlphaFold2가 Nature and Science에 출시되어 생물학 및 AI 학계를 압도했습니다.
1년 후 Meta는 훨씬 더 빠른 ESMFold를 출시했습니다.
빠를 뿐만 아니라 모델에는 150억 개의 매개변수가 있습니다.

LeCun은 트위터를 통해 Meta-FAIR 단백질 팀의 새로운 성과라고 칭찬했습니다.

공동 저자인 Zeming Lin은 30억 개의 매개변수가 있는 대형 모델이 256개의 GPU에서 3주 동안 훈련된 반면 ESMfold는 128개의 GPU에서 10일이 걸렸다고 밝혔습니다. 150억 개의 매개변수 버전에 대해서는 아직 불분명합니다.
코드는 나중에 오픈소스로 공개될 예정이라고 했으니 계속 지켜봐주세요!
크고 빠릅니다!
오늘의 주인공은 개별 단백질 서열로부터 높은 정확도, end-to-end, 원자 수준의 구조를 직접 예측하는 모델인 ESMFold입니다.

논문 주소: https://www.biorxiv.org/content/10.1101/2022.07.20.500902v1
150억 개의 매개변수가 가져오는 이점은 말할 것도 없이 - 학습을 통해 오늘날의 대형 모델은 세 가지를 예측할 수 있습니다. -원자 크기 정확도를 지닌 단백질의 차원 구조.
정확도 측면에서 ESMFold는 AlphaFold2 및 RoseTTAFold와 거의 동일합니다.
하지만 ESMFold의 추론 속도는 AlphaFold2보다 훨씬 빠릅니다!
크기의 순서로 이야기하면 세 가지의 속도를 비교하면 이해하기 어려울 수 있습니다.

차이점은 무엇인가요?
AlphaFold2와 RoseTTAFold는 원자 분해능 구조 예측 문제에서 획기적인 성공을 거두었지만 최적의 성능을 달성하기 위해 다중 서열 정렬(MSA) 및 유사한 단백질 구조 템플릿을 사용합니다.
반면, ESMFold는 언어 모델의 내부 표현을 활용하여 하나의 시퀀스만 입력으로 사용하여 해당 구조 예측을 생성할 수 있으므로 구조 예측 속도가 크게 향상됩니다.

연구원들은 낮은 복잡도 시퀀스에 대한 ESMFold의 예측이 현재의 최첨단 모델과 유사하다는 것을 발견했습니다.
게다가 구조 예측의 정확성은 언어 모델의 복잡성과 밀접한 관련이 있습니다. 즉, 언어 모델이 시퀀스를 더 잘 이해할 수 있을 때 구조를 더 잘 이해할 수 있습니다.

현재 구조와 기능이 알려지지 않은 수십억 개의 단백질 서열이 있으며, 그 중 대부분은 메타게놈 서열 분석에서 파생됩니다.
ESMFold를 사용하여 연구자들은 단 6시간 만에 1백만 개의 메타게놈 서열의 무작위 샘플을 접을 수 있습니다.

이 중 상당수는 신뢰도가 높으며 알려진 구조와 다릅니다(데이터베이스에 기록이 없음).
연구원들은 ESMFold가 현재의 이해를 넘어서는 단백질 구조를 이해하는 데 도움이 될 수 있다고 믿습니다.

또한 ESMFold의 예측은 기존 모델보다 훨씬 빠르기 때문에 연구자들은 ESMFold를 사용하여 빠르게 성장하는 단백질 서열 데이터베이스와 느리게 성장하는 단백질 구조 및 기능 데이터베이스 사이의 격차를 해소하는 데 도움을 줄 수 있습니다.
150억 매개변수 단백질 언어 모델
다음으로 Meta의 새로운 ESMFold에 대해 자세히 이야기해보겠습니다.
ESM-2는 Transformer 기반 언어 모델이며 Attention 메커니즘을 사용하여 입력 시퀀스에서 아미노산 쌍 간의 상호 작용 패턴을 학습합니다.
이전 세대 모델인 ESM-1b와 비교하여 Meta는 모델 구조와 훈련 매개변수를 개선하고 컴퓨팅 리소스와 데이터를 추가했습니다. 동시에 상대 위치 임베딩을 추가하면 모델을 모든 길이의 시퀀스로 일반화할 수 있습니다.
결과에서 1억 5천만 개의 매개변수를 가진 ESM-2 모델이 6억 5천만 개의 매개변수를 가진 ESM-1b 모델보다 더 나은 성능을 보였습니다.
또한 ESM-2는 구조 예측 벤치마크에서도 다른 단백질 언어 모델을 능가합니다. 이러한 성능 향상은 대규모 언어 모델링 분야에서 확립된 패턴과 일치합니다.

ESM-2의 규모가 커질수록 언어 모델링의 정확도가 크게 향상되는 것을 볼 수 있습니다.

종단 간 단일 시퀀스 구조 예측
SMFold와 AlphaFold2의 주요 차이점은 ESMFold가 언어 모델 표현을 사용하므로 명시적인 상동 시퀀스(MSA 형식)가 입력으로 필요하지 않다는 것입니다.
ESMFold는 MSA를 처리하는 계산 비용이 많이 드는 네트워크 모듈을 시퀀스를 처리하는 Transformer 모듈로 대체하여 AlphaFold2의 Evoformer를 단순화합니다. 이러한 단순화는 ESMFold가 MSA 기반 모델보다 훨씬 빠르다는 것을 의미합니다.
접힌 백본의 출력은 최종 원자 수준 구조 및 예측 신뢰도 출력을 담당하는 구조 모듈에 의해 처리됩니다.

연구원들은 CAMEO(2022년 4월~2022년 6월) 및 CASP14(2020년 5월) 테스트 세트에서 ESMFold를 AlphaFold2 및 RoseTTAFold와 비교했습니다.
단일 시퀀스만 입력할 경우 ESMFold는 Alphafold 2보다 훨씬 더 나은 성능을 발휘합니다.
그리고 전체 파이프라인을 사용할 때 AlphaFold2는 CAMEO와 CASP14에서 각각 88.3과 84.7을 달성했습니다. ESMFold는 평균 TM 점수 82.0으로 CAMEO의 RoseTTAfold와 비슷한 정확도를 달성했습니다.

결론
연구자들은 진화적으로 다양한 단백질 서열의 대규모 데이터베이스에서 훈련된 비지도 학습을 목표로 하는 언어 모델이 원자 수준 분해능에서 단백질 구조를 예측할 수 있음을 발견했습니다.
언어 모델의 매개변수를 15B로 확장하면 규모가 단백질 구조 학습에 미치는 영향을 체계적으로 연구할 수 있습니다.
우리는 단백질 구조 예측의 비선형 곡선이 모델 크기의 함수임을 확인하고 언어 모델이 시퀀스를 얼마나 잘 이해하는지와 그 구조 예측 사이의 강한 연관성을 관찰합니다.
ESM-2 시리즈의 모델은 현재까지 훈련된 가장 큰 단백질 언어 모델이며, 최근 개발된 가장 큰 텍스트 모델보다 매개변수가 10배 더 적습니다.
게다가 ESM-2는 이전 모델에 비해 매우 크게 개선되었습니다. 150M 매개변수에서도 ESM-2는 650M 매개변수에서 ESM-1 세대 언어 모델보다 더 정확한 구조 지도를 캡처합니다.
연구원들은 ESMFold 성능의 가장 큰 동인은 언어 모델이라고 밝혔습니다. 언어 모델의 복잡성과 구조 예측의 정확성 사이에는 강한 연관성이 있기 때문에 ESM-2가 단백질 서열을 더 잘 이해할 수 있을 때 현재 최첨단 모델에 필적하는 예측을 달성할 수 있다는 것을 발견했습니다.
ESMFold는 정확한 원자 해상도 구조 예측을 얻었으며 추론 시간은 AlphaFold2보다 훨씬 빠릅니다.
실제로는 속도 이점이 훨씬 더 큽니다. ESMFold는 MSA를 구성하기 위해 진화적으로 관련된 서열을 검색할 필요가 없기 때문입니다.
검색 시간을 줄이는 더 빠른 방법이 있지만 아무리 줄여도 검색 시간이 매우 길어질 수 있습니다.
추론 시간이 크게 단축되어 얻는 이점은 자명합니다. 속도가 향상되면 대규모 메타게놈 서열 데이터베이스의 구조적 공간을 매핑하는 것이 가능해집니다.
원위 상동성과 보존을 식별하는 구조 기반 도구 외에도 ESMFold를 사용한 빠르고 정확한 구조 예측은 대규모의 새로운 서열 컬렉션의 구조적 및 기능적 분석에서 중요한 역할을 할 수 있습니다.
제한된 시간 내에 수백만 개의 예측 구조에 접근하면 천연 단백질의 폭과 다양성에 대한 새로운 통찰력을 발견하고 완전히 새로운 단백질 구조와 단백질 기능을 발견하는 데 도움이 됩니다.
저자 소개
이 글의 공동저자는 Meta AI의 Zeming Lin입니다.

개인 홈페이지에 따르면 Zeming은 뉴욕대학교에서 박사학위를 취득하고 Meta AI에서 연구엔지니어(방문)로 근무하며 주로 백엔드 인프라 작업을 담당했다고 합니다.
그는 버지니아 대학교에서 학사 및 석사 학위를 모두 취득했으며, Qi Yanjun과 함께 기계 학습 응용 분야, 특히 단백질 구조 예측 분야를 연구했습니다.
제 관심분야는 딥러닝, 구조예측, 정보생물학입니다.
위 내용은 0보다 빠르다! Meta는 AlphaFold2를 분쇄하기 위해 150억 개의 매개변수를 갖춘 대규모 단백질 모델을 출시했습니다.의 상세 내용입니다. 자세한 내용은 PHP 중국어 웹사이트의 기타 관련 기사를 참조하세요!

핫 AI 도구

Undresser.AI Undress
사실적인 누드 사진을 만들기 위한 AI 기반 앱

AI Clothes Remover
사진에서 옷을 제거하는 온라인 AI 도구입니다.

Undress AI Tool
무료로 이미지를 벗다

Clothoff.io
AI 옷 제거제

AI Hentai Generator
AI Hentai를 무료로 생성하십시오.

인기 기사
뜨거운 도구

메모장++7.3.1
사용하기 쉬운 무료 코드 편집기

SublimeText3 중국어 버전
중국어 버전, 사용하기 매우 쉽습니다.

스튜디오 13.0.1 보내기
강력한 PHP 통합 개발 환경

드림위버 CS6
시각적 웹 개발 도구

SublimeText3 Mac 버전
신 수준의 코드 편집 소프트웨어(SublimeText3)
뜨거운 주제
 7475
7475
 15
1377
52
77
11
49
19
19
31
15
1377
52
77
11
49
19
19
31

 딥마인드 로봇이 탁구를 치는데 포핸드와 백핸드가 공중으로 미끄러져 인간 초보자를 완전히 제압했다.
Aug 09, 2024 pm 04:01 PM
딥마인드 로봇이 탁구를 치는데 포핸드와 백핸드가 공중으로 미끄러져 인간 초보자를 완전히 제압했다.
Aug 09, 2024 pm 04:01 PM
하지만 공원에 있는 노인을 이길 수는 없을까요? 파리올림픽이 본격화되면서 탁구가 많은 주목을 받고 있다. 동시에 로봇은 탁구 경기에서도 새로운 돌파구를 마련했습니다. 방금 DeepMind는 탁구 경기에서 인간 아마추어 선수 수준에 도달할 수 있는 최초의 학습 로봇 에이전트를 제안했습니다. 논문 주소: https://arxiv.org/pdf/2408.03906 DeepMind 로봇은 탁구를 얼마나 잘 치나요? 아마도 인간 아마추어 선수들과 동등할 것입니다: 포핸드와 백핸드 모두: 상대는 다양한 플레이 스타일을 사용하고 로봇도 견딜 수 있습니다: 다양한 스핀으로 서브를 받습니다. 그러나 게임의 강도는 그만큼 강렬하지 않은 것 같습니다. 공원에 있는 노인. 로봇용, 탁구용
 하나의 기사로 토큰화를 이해해보세요!
Apr 12, 2024 pm 02:31 PM
하나의 기사로 토큰화를 이해해보세요!
Apr 12, 2024 pm 02:31 PM
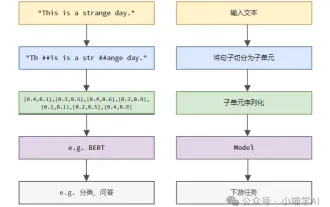
언어 모델은 일반적으로 문자열 형식인 텍스트에 대해 추론하지만 모델에 대한 입력은 숫자만 가능하므로 텍스트를 숫자 형식으로 변환해야 합니다. 토큰화는 자연어 처리의 기본 작업으로, 연속적인 텍스트 시퀀스(예: 문장, 단락 등)를 특정 필요에 따라 문자 시퀀스(예: 단어, 구, 문자, 구두점 등)로 나눌 수 있습니다. 그 안에 있는 단위를 토큰 또는 단어라고 합니다. 아래 그림에 표시된 특정 프로세스에 따르면 먼저 텍스트 문장을 단위로 나눈 다음 단일 요소를 디지털화(벡터로 매핑)한 다음 이러한 벡터를 인코딩 모델에 입력하고 마지막으로 다운스트림 작업으로 출력하여 다음 작업을 수행합니다. 추가로 최종 결과를 얻으십시오. 텍스트 분할은 텍스트 분할의 세분성에 따라 Toke로 나눌 수 있습니다.
 대형 모델에 대한 새로운 과학적이고 복잡한 질문 답변 벤치마크 및 평가 시스템을 제공하기 위해 UNSW, Argonne, University of Chicago 및 기타 기관이 공동으로 SciQAG 프레임워크를 출시했습니다.
Jul 25, 2024 am 06:42 AM
대형 모델에 대한 새로운 과학적이고 복잡한 질문 답변 벤치마크 및 평가 시스템을 제공하기 위해 UNSW, Argonne, University of Chicago 및 기타 기관이 공동으로 SciQAG 프레임워크를 출시했습니다.
Jul 25, 2024 am 06:42 AM
편집자 |ScienceAI 질문 응답(QA) 데이터 세트는 자연어 처리(NLP) 연구를 촉진하는 데 중요한 역할을 합니다. 고품질 QA 데이터 세트는 모델을 미세 조정하는 데 사용될 수 있을 뿐만 아니라 LLM(대형 언어 모델)의 기능, 특히 과학적 지식을 이해하고 추론하는 능력을 효과적으로 평가하는 데에도 사용할 수 있습니다. 현재 의학, 화학, 생물학 및 기타 분야를 포괄하는 과학적인 QA 데이터 세트가 많이 있지만 이러한 데이터 세트에는 여전히 몇 가지 단점이 있습니다. 첫째, 데이터 형식이 비교적 단순하고 대부분이 객관식 질문이므로 평가하기 쉽지만 모델의 답변 선택 범위가 제한되고 모델의 과학적 질문 답변 능력을 완전히 테스트할 수 없습니다. 이에 비해 개방형 Q&A는
 클라우드에 대규모 모델을 배포하기 위한 세 가지 비밀
Apr 24, 2024 pm 03:00 PM
클라우드에 대규모 모델을 배포하기 위한 세 가지 비밀
Apr 24, 2024 pm 03:00 PM
편집|제작자 Xingxuan|51CTO 기술 스택(WeChat ID: blog51cto) 지난 2년 동안 저는 기존 시스템보다는 대규모 언어 모델(LLM)을 사용하는 생성 AI 프로젝트에 더 많이 참여해 왔습니다. 서버리스 클라우드 컴퓨팅이 그리워지기 시작했습니다. 이들의 애플리케이션은 대화형 AI 강화부터 다양한 산업에 대한 복잡한 분석 솔루션 제공 및 기타 다양한 기능에 이르기까지 다양합니다. 퍼블릭 클라우드 제공업체가 이미 기성 생태계를 제공하고 있으며 이것이 저항이 가장 적은 경로이기 때문에 많은 기업이 이러한 모델을 클라우드 플랫폼에 배포합니다. 그러나 저렴하지는 않습니다. 클라우드는 확장성, 효율성, 고급 컴퓨팅 기능(요청 시 GPU 사용 가능)과 같은 다른 이점도 제공합니다. 퍼블릭 클라우드 플랫폼에 LLM을 배포하는 프로세스에는 잘 알려지지 않은 몇 가지 측면이 있습니다.
 대규모 언어 모델의 효율적인 매개변수 미세 조정--BitFit/Prefix/Prompt 미세 조정 시리즈
Oct 07, 2023 pm 12:13 PM
대규모 언어 모델의 효율적인 매개변수 미세 조정--BitFit/Prefix/Prompt 미세 조정 시리즈
Oct 07, 2023 pm 12:13 PM
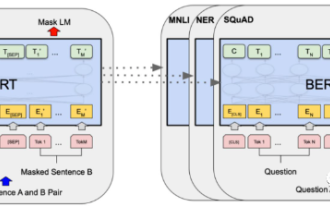
2018년 Google은 BERT를 출시한 후 11개 NLP 작업의 State-of-the-art(Sota) 결과를 단번에 무너뜨리며 NLP 세계의 새로운 이정표가 되었습니다. 아래 그림에서 왼쪽은 BERT 모델 사전 설정이고 오른쪽은 특정 작업에 대한 미세 조정 프로세스입니다. 그중 미세 조정 단계는 텍스트 분류, 품사 태깅, 질문 및 답변 시스템 등과 같은 일부 다운스트림 작업에서 이후에 사용될 때 미세 조정을 위한 것입니다. BERT는 다양한 환경에서 미세 조정할 수 있습니다. 구조를 조정하지 않고 작업을 수행합니다. "사전 학습된 언어 모델 + 다운스트림 작업 미세 조정" 작업 설계를 통해 강력한 모델 효과를 제공합니다. 이후 '사전 학습 언어 모델 + 다운스트림 작업 미세 조정'이 NLP 분야의 주류 학습이 되었습니다.
 단백질과 모든 살아있는 분자의 상호 작용과 구조를 이전보다 훨씬 더 정확하게 예측하는 AlphaFold 3 출시
Jul 16, 2024 am 12:08 AM
단백질과 모든 살아있는 분자의 상호 작용과 구조를 이전보다 훨씬 더 정확하게 예측하는 AlphaFold 3 출시
Jul 16, 2024 am 12:08 AM
Editor | Radish Skin 2021년 강력한 AlphaFold2가 출시된 이후 과학자들은 단백질 구조 예측 모델을 사용하여 세포 내 다양한 단백질 구조를 매핑하고 약물을 발견하며 알려진 모든 단백질 상호 작용에 대한 "우주 지도"를 그려 왔습니다. 방금 Google DeepMind는 단백질, 핵산, 소분자, 이온 및 변형된 잔기를 포함한 복합체에 대한 결합 구조 예측을 수행할 수 있는 AlphaFold3 모델을 출시했습니다. AlphaFold3의 정확도는 과거의 많은 전용 도구(단백질-리간드 상호작용, 단백질-핵산 상호작용, 항체-항원 예측)에 비해 크게 향상되었습니다. 이는 단일 통합 딥러닝 프레임워크 내에서 다음을 달성할 수 있음을 보여줍니다.
 RoSA: 대규모 모델 매개변수를 효율적으로 미세 조정하기 위한 새로운 방법
Jan 18, 2024 pm 05:27 PM
RoSA: 대규모 모델 매개변수를 효율적으로 미세 조정하기 위한 새로운 방법
Jan 18, 2024 pm 05:27 PM
언어 모델이 전례 없는 규모로 확장됨에 따라 다운스트림 작업에 대한 포괄적인 미세 조정 비용이 엄청나게 높아집니다. 이러한 문제를 해결하기 위해 연구자들은 PEFT 방식에 주목하고 채택하기 시작했다. PEFT 방법의 주요 아이디어는 미세 조정 범위를 작은 매개변수 세트로 제한하여 계산 비용을 줄이면서도 자연어 이해 작업에서 최첨단 성능을 달성하는 것입니다. 이러한 방식으로 연구자들은 고성능을 유지하면서 컴퓨팅 리소스를 절약할 수 있어 자연어 처리 분야에 새로운 연구 핫스팟을 가져올 수 있습니다. RoSA는 일련의 벤치마크에 대한 실험을 통해 동일한 매개변수 예산을 사용하는 이전 LoRA(낮은 순위 적응형) 및 순수 희소 미세 조정 방법보다 성능이 뛰어난 것으로 밝혀진 새로운 PEFT 기술입니다. 이 기사에서는 심층적으로 다룰 것입니다.
 역사상 가장 큰 ViT를 편리하게 훈련하셨나요? Google, 시각적 언어 모델 PaLI 업그레이드: 100개 이상의 언어 지원
Apr 12, 2023 am 09:31 AM
역사상 가장 큰 ViT를 편리하게 훈련하셨나요? Google, 시각적 언어 모델 PaLI 업그레이드: 100개 이상의 언어 지원
Apr 12, 2023 am 09:31 AM
최근 몇 년간 자연어 처리의 발전은 주로 대규모 언어 모델에서 비롯되었습니다. 출시되는 각각의 새로운 모델은 매개변수와 훈련 데이터의 양을 새로운 최고치로 끌어올리는 동시에 기존 벤치마크 순위를 무너뜨릴 것입니다. 예를 들어, 올해 4월 Google은 5,400억 매개변수의 언어 모델 PaLM(Pathways Language Model)을 출시했는데, 이는 일련의 언어 및 추론 테스트에서 인간을 성공적으로 능가했으며, 특히 소수의 소규모 샘플 학습 시나리오에서 탁월한 성능을 발휘했습니다. PaLM은 차세대 언어 모델의 개발 방향으로 간주됩니다. 마찬가지로 시각적 언어 모델은 실제로 놀라운 효과를 발휘하며 모델의 크기를 늘려 성능을 향상할 수 있습니다. 물론 멀티 태스킹 시각적 언어 모델에 불과하다면
