

기하학적 딥러닝 방법을 사용하여 약물 분자 합성을 위한 최적 옵션을 예측하여 신약 발견의 길을 열었습니다.


후기능화는 약물 후보물질의 특성을 최적화하는 경제적인 방법입니다. 그러나 약물 분자의 화학적 복잡성으로 인해 종종 후기 단계 기능화가 어려워집니다.
이 문제를 해결하기 위해 뮌헨 대학, ETH Zurich 및 Roche Basel의 연구자들은 협력하여 후기 단계 기능화 플랫폼을 기반으로 합니다. 기하학적 딥 러닝 및 고처리량 반응 스크리닝 기술
보릴화는 기능화의 핵심 단계 중 하나라는 점을 고려하여 계산 모델을 사용하여 평균 절대 오차 범위가 4~5%인 다양한 반응 조건에서 수율을 예측했습니다. 이 모델은 각각 92%와 67%의 정확도로 알려진 기질과 알려지지 않은 기질에 대한 새로운 반응을 분류할 수 있었습니다. 우리는 분류기의 F 점수가 67%로 주요 제품의 위치 선택성을 정확하게 포착할 수 있었습니다. 23개의 다양한 상업용 약물 분자에 적용했을 때 우리는 구조적 다양화를 위한 많은 기회를 성공적으로 발견했습니다
이 연구는 "기하학적 딥러닝을 사용하여 고처리량 실험을 활성화하여 후기 단계 약물 다양화 촉진"이라는 제목으로 2023년 11월에 발표되었습니다. 3월 23일 Nature Chemistry 저널에 게재됨

LSF 프로젝트는 의약 화학 연구에서 중요한 역할을 합니다.
의약 화학에서 구조-활성 관계를 확립하는 것을 목표로 할 때 구조의 참신함과 복잡성으로 인해 화학 표적 구조 합성이 어려워집니다. 구조-활성 관계 모델은 선도 화합물 및 선도 화합물 최적화 계획을 안내하여 약물 후보의 약리학적 활성 및 물리화학적 특성을 개선할 수 있습니다. 구조-활동 관계를 탐색하려면 설계-제작-테스트-분석 주기의 병목 현상인 효율적인 통합이 중요합니다.
유기 지지체의 후기 단계 기능화를 위해 C-H 결합을 활성화하고 수정하는 많은 대체 방법이 있습니다( LSF)는 분자 빌딩 블록부터 고급 제약 분자까지 다양합니다. 많은 촉매 시스템은 변형된 유사체에 대한 화학적 및 위치 선택적 접근뿐만 아니라 방향성 및 비방향성 접근 방식을 제공합니다.
수많은 LSF 방법 중에서 C-H 보릴화 방법은 신속한 화합물 다양화를 위해 가장 일반적으로 사용되는 방법으로 간주됩니다. 유기 붕소 화합물은 후속 C-C 결합 커플링 반응을 위한 신뢰할 수 있는 수단으로 다양한 작용기로 전환될 수 있으므로 광범위한 구조-활성 관계 연구가 가능합니다
그러나 현재 약물 발견 보고서에서 LSF의 적용은 소수에 불과합니다. 이러한 보고서의 대부분은 단일 LSF 반응 유형에 중점을 둡니다. 서로 다른 결합 강도, 전자 특성, 입체 및 작용기 환경을 갖는 여러 유형의 C-H 결합의 직접 LSF는 문제를 야기합니다. 더욱이, LSF 프로젝트를 수행하는 것은 종종 시간과 자원 집약적이며, 이는 많은 의약 화학 프로젝트의 빡빡한 일정과 제한된 자산과 일치하지 않습니다
이 차트는 붕소화 다양화 연구의 개요를 보여줍니다. (데이터 출처: 논문)
인공지능 기반 LSF(언어 지원 기능)
고처리량 실험(HTE)은 반자동 소형화 및 소량 배치 스크리닝이 가능한 확립된 반응 최적화 방법으로, 소수의 귀중한 빌딩 블록과 소모품을 사용하여 여러 변환을 동시에 재현 가능하게 실행합니다. 성공 및 실패 응답에 대한 고품질 데이터세트를 생성하는 FAIR(검색 가능성, 접근성, 상호 운용성, 재사용성) 문서와 결합된 HTE는 고급 데이터 분석 및 기계 학습을 통해 약물 발견을 위한 LSF를 실현합니다.
GNN(Graph Neural Network)은 분자 특징 추출 및 속성 예측에 널리 사용되었습니다. 화학 반응 계획을 위해 개발된 다양한 기계 학습 방법 중에서 GNN은 역합성 계획, 위치 선택성 예측 및 반응 생성물 예측에 성공적으로 적용되었습니다. 또한 비슷한 문제를 해결하기 위해 변환기와 지문 기반 방법도 개발되었습니다.
연구에 따르면 전이 상태의 기하학을 학습하면 경쟁 반응의 결과를 정확하게 예측할 수 있는 것으로 나타났습니다. 밀도 범함수 이론(DFT)과 원자 부분 전하의 그래픽 특성화를 사용하면 전자 효과에 의해 구동되는 반응의 위치 선택성에 대한 예측이 향상될 수 있습니다. 그래프 기계 학습과 고처리량 실험(HTE)을 결합하여 유기 기판의 C-H 활성화 반응 조건을 최적화할 수 있습니다. 일부 연구에서는 거울상 선택성을 포함하여 반응 결과를 예측할 수 있는 전이 상태의 딥 러닝 모델 사용에 초점을 맞췄습니다.
그러나 이러한 방법은 작은 분자 구조와 상대적으로 작은 데이터 세트로 제한되어 이러한 모델을 구조적으로 더 복잡한 약물 유사 분자에 적용합니다. 문헌 연구를 기반으로 이리듐 촉매 보릴화 반응의 위치 선택성은 전이 상태의 양자 화학 정보로 강화된 하이브리드 기계 학습 모델을 통해 예측할 수 있습니다. 그러나 C-H 활성화 반응 모델의 성능과 다중 방향족 고리 시스템을 가진 분자에서의 위치 선택적 적용에 대한 입체 효과와 전자 효과의 영향은 아직 연구되지 않았습니다
기하학적 딥러닝을 사용한 자동화된 LSF 붕소화 스크리닝
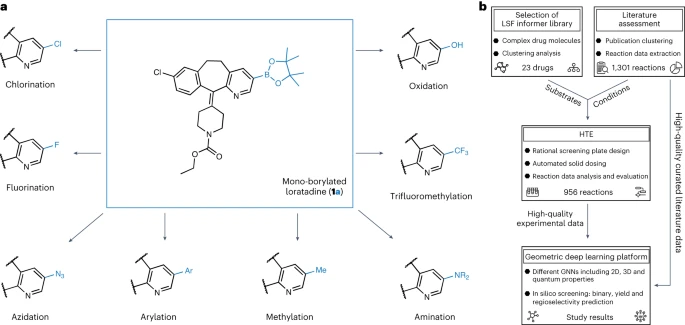
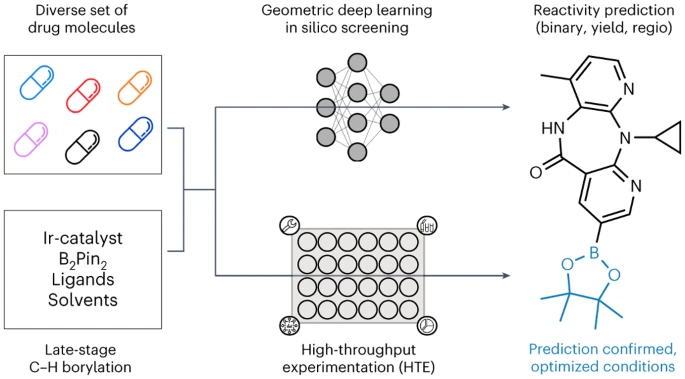
뮌헨 대학, ETH Zurich, Roche Pharmaceuticals Basel의 연구원들은 후기 단계의 히트 및 리드 다양화 기회를 식별하기 위해 기하학적 딥러닝에 적용되는 자동화된 LSF 붕소화 스크리닝 방법을 소개합니다. 복잡한 약물 분자 LSF의 반응 결과, 수율 및 위치 선택성을 예측하기 위해 컴퓨터 딥 러닝을 사용했습니다.
LMU 화학 및 약학대학과 Roche 연구 그룹의 수석 저자이자 박사 과정 학생인 David Nippa는 이 접근 방식을 사용하면 실험실 실험 횟수를 크게 줄여서 효율성을 높일 수 있다고 말했습니다. 화학적 합성 및 지속 가능성
이 연구의 첫 번째 단계에서 우리는 후기 단계 약물 발견 선도 화합물의 특성과 관련된 적절한 고처리량 스크리닝 반응 조건 및 기질을 선택하기 위해 출판된 문헌을 철저히 분석했습니다. 우리는 수동으로 정리된 38개의 문헌 데이터 세트를 기반으로 반응 조건을 결정했습니다
LSF 기질의 선택은 1,174개의 승인된 약물에 대한 클러스터 분석 결과를 기반으로 하여 구조적으로 다른 23개의 약물 분자를 생성했습니다. 이 접근 방식을 통해 연구자들은 납 화합물 합성을 최적화하기 위해 이상적인 기질과 적용 가능성이 제한된 단편에만 의존하는 대신 "정보 라이브러리" 접근 방식에서 반응 조건 및 기질의 관련 사례를 사용할 수 있습니다.
두 번째 단계에서 연구자들은 세미 -데이터(실험 데이터세트)를 생성하기 위한 자동화된 고처리량 실험(HTE). 선택된 약물 분자 및 반응 조건의 반응 데이터는 후속 반응 결과의 기계 학습을 위한 고품질 데이터를 제공합니다
마지막으로 2차원, 3차원 및 원자 부분 전하를 사용하여 다양한 그래프 신경망(GNN) 모델을 훈련했습니다. 강화 분자 그래프를 사용하여 이원 반응 결과, 반응 수율 및 위치 선택성을 예측합니다. ETH Zurich의 박사과정 학생인 Kenneth Atz는 "흥미롭게도 출발 물질에 대한 2차원 화학 공식이 아닌 3차원 정보를 고려할 때 예측이 향상되었습니다."라고 말했습니다.
이 접근 방식은 위치를 식별하는 데 성공적으로 사용되었습니다. 추가적인 반응성 그룹이 도입될 수 있는 기존 활성 성분 내에서. 이는 연구자들이 알려진 제약 활성 성분의 새롭고 더욱 효과적인 변종을 더욱 신속하게 개발하는 데 도움이 될 것입니다.
논문 내용을 보려면 다음 링크를 클릭하십시오: https://www.nature.com/articles/s41557-023-01360-5
관련 보고서: https://techxplore.com/news/2023-11-인공 지능이 약물의 길을 열어줍니다.html
위 내용은 기하학적 딥러닝 방법을 사용하여 약물 분자 합성을 위한 최적 옵션을 예측하여 신약 발견의 길을 열었습니다.의 상세 내용입니다. 자세한 내용은 PHP 중국어 웹사이트의 기타 관련 기사를 참조하세요!

핫 AI 도구

Undresser.AI Undress
사실적인 누드 사진을 만들기 위한 AI 기반 앱

AI Clothes Remover
사진에서 옷을 제거하는 온라인 AI 도구입니다.

Undress AI Tool
무료로 이미지를 벗다

Clothoff.io
AI 옷 제거제

AI Hentai Generator
AI Hentai를 무료로 생성하십시오.

인기 기사
뜨거운 도구

메모장++7.3.1
사용하기 쉬운 무료 코드 편집기

SublimeText3 중국어 버전
중국어 버전, 사용하기 매우 쉽습니다.

스튜디오 13.0.1 보내기
강력한 PHP 통합 개발 환경

드림위버 CS6
시각적 웹 개발 도구

SublimeText3 Mac 버전
신 수준의 코드 편집 소프트웨어(SublimeText3)
뜨거운 주제
 7510
7510
 15
1378
52
78
11
53
19
19
64
15
1378
52
78
11
53
19
19
64

 딥마인드 로봇이 탁구를 치는데 포핸드와 백핸드가 공중으로 미끄러져 인간 초보자를 완전히 제압했다.
Aug 09, 2024 pm 04:01 PM
딥마인드 로봇이 탁구를 치는데 포핸드와 백핸드가 공중으로 미끄러져 인간 초보자를 완전히 제압했다.
Aug 09, 2024 pm 04:01 PM
하지만 공원에 있는 노인을 이길 수는 없을까요? 파리올림픽이 본격화되면서 탁구가 많은 주목을 받고 있다. 동시에 로봇은 탁구 경기에서도 새로운 돌파구를 마련했습니다. 방금 DeepMind는 탁구 경기에서 인간 아마추어 선수 수준에 도달할 수 있는 최초의 학습 로봇 에이전트를 제안했습니다. 논문 주소: https://arxiv.org/pdf/2408.03906 DeepMind 로봇은 탁구를 얼마나 잘 치나요? 아마도 인간 아마추어 선수들과 동등할 것입니다: 포핸드와 백핸드 모두: 상대는 다양한 플레이 스타일을 사용하고 로봇도 견딜 수 있습니다: 다양한 스핀으로 서브를 받습니다. 그러나 게임의 강도는 그만큼 강렬하지 않은 것 같습니다. 공원에 있는 노인. 로봇용, 탁구용
 최초의 기계식 발톱! Yuanluobao는 2024년 세계 로봇 회의에 등장하여 집에 들어갈 수 있는 최초의 체스 로봇을 출시했습니다.
Aug 21, 2024 pm 07:33 PM
최초의 기계식 발톱! Yuanluobao는 2024년 세계 로봇 회의에 등장하여 집에 들어갈 수 있는 최초의 체스 로봇을 출시했습니다.
Aug 21, 2024 pm 07:33 PM
8월 21일, 2024년 세계로봇대회가 베이징에서 성대하게 개최되었습니다. SenseTime의 홈 로봇 브랜드 "Yuanluobot SenseRobot"은 전체 제품군을 공개했으며, 최근에는 Yuanluobot AI 체스 두는 로봇인 체스 프로페셔널 에디션(이하 "Yuanluobot SenseRobot")을 출시하여 세계 최초의 A 체스 로봇이 되었습니다. 집. Yuanluobo의 세 번째 체스 게임 로봇 제품인 새로운 Guoxiang 로봇은 AI 및 엔지니어링 기계 분야에서 수많은 특별한 기술 업그레이드와 혁신을 거쳤으며 처음으로 3차원 체스 말을 집는 능력을 실현했습니다. 가정용 로봇의 기계 발톱을 통해 체스 게임, 모두 체스 게임, 기보 복습 등과 같은 인간-기계 기능을 수행합니다.
 클로드도 게으르게 됐어요! 네티즌 : 휴가를 보내는 법을 배우십시오
Sep 02, 2024 pm 01:56 PM
클로드도 게으르게 됐어요! 네티즌 : 휴가를 보내는 법을 배우십시오
Sep 02, 2024 pm 01:56 PM
개학이 코앞으로 다가왔습니다. 새 학기를 앞둔 학생들뿐만 아니라 대형 AI 모델도 스스로 관리해야 합니다. 얼마 전 레딧에는 클로드가 게으르다고 불평하는 네티즌들이 붐볐습니다. "레벨이 많이 떨어졌고, 자주 멈췄고, 심지어 출력도 매우 짧아졌습니다. 출시 첫 주에는 4페이지 전체 문서를 한 번에 번역할 수 있었지만 지금은 반 페이지도 출력하지 못합니다. !" https://www.reddit.com/r/ClaudeAI/comments/1by8rw8/something_just_feels_wrong_with_claude_in_the/ "클로드에게 완전히 실망했습니다"라는 제목의 게시물에
 세계로봇컨퍼런스에서 '미래 노인돌봄의 희망'을 담은 국산 로봇이 포위됐다.
Aug 22, 2024 pm 10:35 PM
세계로봇컨퍼런스에서 '미래 노인돌봄의 희망'을 담은 국산 로봇이 포위됐다.
Aug 22, 2024 pm 10:35 PM
베이징에서 열린 세계로봇컨퍼런스에서는 휴머노이드 로봇의 전시가 현장의 절대 화두가 됐다. 스타더스트 인텔리전트 부스에서는 AI 로봇 어시스턴트 S1이 덜시머, 무술, 서예 3대 퍼포먼스를 선보였다. 문학과 무술을 모두 갖춘 하나의 전시 공간은 수많은 전문 관객과 미디어를 끌어 모았습니다. 탄력 있는 현의 우아한 연주를 통해 S1은 정밀한 작동과 속도, 힘, 정밀성을 갖춘 절대적인 제어력을 보여줍니다. CCTV 뉴스는 '서예'의 모방 학습 및 지능형 제어에 대한 특별 보도를 진행했습니다. 회사 설립자 Lai Jie는 부드러운 움직임 뒤에 하드웨어 측면이 최고의 힘 제어와 가장 인간과 유사한 신체 지표(속도, 하중)를 추구한다고 설명했습니다. 등)이지만 AI측에서는 사람의 실제 움직임 데이터를 수집해 로봇이 강한 상황에 직면했을 때 더욱 강해지고 빠르게 진화하는 방법을 학습할 수 있다. 그리고 민첩하다
 ACL 2024 시상식 발표: HuaTech의 Oracle 해독에 관한 최고의 논문 중 하나, GloVe Time Test Award
Aug 15, 2024 pm 04:37 PM
ACL 2024 시상식 발표: HuaTech의 Oracle 해독에 관한 최고의 논문 중 하나, GloVe Time Test Award
Aug 15, 2024 pm 04:37 PM
참가자들은 이번 ACL 컨퍼런스에서 많은 것을 얻었습니다. ACL2024는 6일간 태국 방콕에서 개최됩니다. ACL은 전산언어학 및 자연어 처리 분야 최고의 국제학술대회로 국제전산언어학회(International Association for Computational Linguistics)가 주최하고 매년 개최된다. ACL은 NLP 분야에서 학술 영향력 1위를 항상 차지하고 있으며, CCF-A 추천 컨퍼런스이기도 합니다. 올해로 62회째를 맞이하는 ACL 컨퍼런스에는 NLP 분야의 최신 저서가 400편 이상 접수됐다. 어제 오후 컨퍼런스에서는 최우수 논문과 기타 상을 발표했습니다. 이번에 최우수논문상 7개(미출판 2개), 우수주제상 1개, 우수논문상 35개가 있다. 이 컨퍼런스에서는 또한 3개의 리소스 논문상(ResourceAward)과 사회적 영향상(Social Impact Award)을 수상했습니다.
 홍멍 스마트 트래블 S9과 풀시나리오 신제품 출시 컨퍼런스, 다수의 블록버스터 신제품이 함께 출시됐다
Aug 08, 2024 am 07:02 AM
홍멍 스마트 트래블 S9과 풀시나리오 신제품 출시 컨퍼런스, 다수의 블록버스터 신제품이 함께 출시됐다
Aug 08, 2024 am 07:02 AM
오늘 오후 Hongmeng Zhixing은 공식적으로 새로운 브랜드와 신차를 환영했습니다. 8월 6일, Huawei는 Hongmeng Smart Xingxing S9 및 Huawei 전체 시나리오 신제품 출시 컨퍼런스를 개최하여 파노라마식 스마트 플래그십 세단 Xiangjie S9, 새로운 M7Pro 및 Huawei novaFlip, MatePad Pro 12.2인치, 새로운 MatePad Air, Huawei Bisheng을 선보였습니다. 레이저 프린터 X1 시리즈, FreeBuds6i, WATCHFIT3 및 스마트 스크린 S5Pro를 포함한 다양한 새로운 올-시나리오 스마트 제품, 스마트 여행, 스마트 오피스, 스마트 웨어에 이르기까지 화웨이는 풀 시나리오 스마트 생태계를 지속적으로 구축하여 소비자에게 스마트한 경험을 제공합니다. 만물인터넷. Hongmeng Zhixing: 스마트 자동차 산업의 업그레이드를 촉진하기 위한 심층적인 권한 부여 화웨이는 중국 자동차 산업 파트너와 손을 잡고
 Li Feifei 팀은 로봇에 공간 지능을 제공하고 GPT-4o를 통합하기 위해 ReKep을 제안했습니다.
Sep 03, 2024 pm 05:18 PM
Li Feifei 팀은 로봇에 공간 지능을 제공하고 GPT-4o를 통합하기 위해 ReKep을 제안했습니다.
Sep 03, 2024 pm 05:18 PM
비전과 로봇 학습의 긴밀한 통합. 최근 화제를 모으고 있는 1X 휴머노이드 로봇 네오(NEO)와 두 개의 로봇 손이 원활하게 협력해 옷 개기, 차 따르기, 신발 싸기 등을 하는 모습을 보면 마치 로봇 시대로 접어들고 있다는 느낌을 받을 수 있다. 실제로 이러한 부드러운 움직임은 첨단 로봇 기술 + 정교한 프레임 디자인 + 다중 모드 대형 모델의 산물입니다. 우리는 유용한 로봇이 종종 환경과 복잡하고 절묘한 상호작용을 요구한다는 것을 알고 있으며, 환경은 공간적, 시간적 영역에서 제약으로 표현될 수 있습니다. 예를 들어, 로봇이 차를 따르도록 하려면 먼저 로봇이 찻주전자 손잡이를 잡고 차를 흘리지 않고 똑바로 세운 다음, 주전자 입구와 컵 입구가 일치할 때까지 부드럽게 움직여야 합니다. 을 누른 다음 주전자를 특정 각도로 기울입니다. 이것
 분산 인공지능 컨퍼런스 DAI 2024 Call for Papers: Agent Day, 강화학습의 아버지 Richard Sutton이 참석합니다! Yan Shuicheng, Sergey Levine 및 DeepMind 과학자들이 기조 연설을 할 예정입니다.
Aug 22, 2024 pm 08:02 PM
분산 인공지능 컨퍼런스 DAI 2024 Call for Papers: Agent Day, 강화학습의 아버지 Richard Sutton이 참석합니다! Yan Shuicheng, Sergey Levine 및 DeepMind 과학자들이 기조 연설을 할 예정입니다.
Aug 22, 2024 pm 08:02 PM
컨퍼런스 소개 과학기술의 급속한 발전과 함께 인공지능은 사회 발전을 촉진하는 중요한 힘이 되었습니다. 이 시대에 우리는 분산인공지능(DAI)의 혁신과 적용을 목격하고 참여할 수 있어 행운입니다. 분산 인공지능(Distributed Artificial Intelligence)은 인공지능 분야의 중요한 한 분야로, 최근 몇 년간 점점 더 많은 주목을 받고 있습니다. 대규모 언어 모델(LLM) 기반 에이전트가 갑자기 등장했습니다. 대규모 모델의 강력한 언어 이해와 생성 기능을 결합하여 자연어 상호 작용, 지식 추론, 작업 계획 등에 큰 잠재력을 보여주었습니다. AIAgent는 빅 언어 모델을 이어받아 현재 AI계에서 화제가 되고 있습니다. 오
