

快了一個0! Meta祭出150億參數蛋白質大模型,碾壓AlphaFold2

迄今為止規模最大的蛋白質語言模型問世了!
一年前,DeepMind開源AlphaFold2連登Nature、Science,刷爆生物和AI學界。
一年後,Meta帶著速度快一個數量級的ESMFold來了。
不光速度快,模型還足足有150億個參數。

LeCun推文稱讚,這是Meta-FAIR蛋白質團隊的偉大新成果。

共同一作Zeming Lin透露,30億參數的大模型在256個GPU上訓練了3個星期,而ESMfold在128個GPU上用了10天。至於150億參數的版本,目前還不清楚。
他也表示,程式碼隨後肯定會開源,敬請關注!
又大又快!
今天,我們的主角是ESMFold,一個從蛋白質個體的序列,直接進行高準確度、端對端、原子層級結構預測的模型。

論文網址:https://www.biorxiv.org/content/10.1101/2022.07.20.500902v1
150億參數帶來的好處不必多說——透過訓練,如今的大模型可以在原子大小的精確度上預測蛋白質的三維結構。
從準確度來看,ESMFold和AlphaFold2、RoseTTAFold差不多。
但是,ESMFold推測速度比AlphaFold2快一個數量級!
一下說數量級可能不好理解三者之間速度的對比,看看下面這張圖就懂了。

有啥差別?
雖然說AlphaFold2和RoseTTAFold在原子解析度結構預測問題上取得了突破性的成功,但它們也依賴於使用多序列比對(MSA)和類似的蛋白質結構模板來實現最佳性能。
相較之下,透過利用語言模型的內部表徵,ESMFold只用一個序列作為輸入就能產生對應的結構預測,從而大大加快了結構預測的速度。

研究人員發現,ESMFold對低複雜度序列的預測與當下最先進的模型相當。
而且,結構預測的準確性與語言模型的複雜度息息相關,也就是說,當語言模型能更好地理解序列時,便可以更好地理解結構。

目前,有數十億結構和功能未知的蛋白質序列,其中許多來自元基因組定序。
利用ESMFold,研究人員只需6小時,就能摺疊完成100萬個元基因組序列的隨機樣本。

其中很大一部分具有高置信度,並且與任何已知的結構不同(在資料庫中沒有記錄)。
研究人員認為,ESMFold可以幫助理解那些超越現有認知的蛋白質結構。

此外,由於ESMFold的預測速度比現有的模型快一個數量級,因此研究人員便可藉助ESMFold來協助填補快速增長的蛋白質序列資料庫與進展緩慢的蛋白質結構和功能資料庫之間的鴻溝。
150億參數的蛋白質語言模型
接下來我們就來具體說Meta這款全新的ESMFold。
ESM-2是一個基於Transformer的語言模型,並使用注意力機制來學習輸入序列中成對胺基酸之間的相互作用模式。
相對於上一代模型ESM-1b,Meta對模型結構、訓練參數進行了改進,並增加了計算資源和資料。同時,相對位置嵌入的加入,使模型能夠推廣到任意長度的序列。
從結果來看,具有1.5億個參數的ESM-2模型比具有6.5億個參數的ESM-1b模型表現得更好。
此外,在結構預測的基準上,ESM-2也超過了其他的蛋白質語言模型。這種表現的提升與大型語言建模領域所建立的規律是一致的。

隨著ESM-2規模的增加,可以觀察到語言建模的精確度有很大的提升。

端對端的單序列結構預測
SMFold和AlphaFold2的一個關鍵區別是,ESMFold使用語言模型表示,消除了對明確的同源序列(以MSA的形式)作為輸入的需要。
ESMFold透過用一個處理序列的Transformer模組取代處理MSA的計算昂貴的網路模組,簡化了AlphaFold2中的Evoformer。這種簡化意味著ESMFold的速度大大提高,遠高於基於MSA的模型。
折疊主幹的輸出接下來又被一個結構模組處理,它負責輸出最終的原子級結構和預測的置信度。

研究人員將ESMFold與AlphaFold2和RoseTTAFold在CAMEO(2022年4月至2022年6月)和CASP14(2020年5月)測試集上進行比較。
當只給單一序列輸入時,ESMFold的表現比Alphafold 2好得多。
而當使用完整的管道時,AlphaFold2在CAMEO和CASP14上分別達到了88.3和84.7。 ESMFold在CAMEO上取得了與RoseTTAfold相當的準確率,其平均TM分數為82.0。

結論
#研究人員發現,以無監督學習為目標的語言模型在一個大型的進化多樣化的蛋白質序列資料庫中訓練,能夠對蛋白質結構進行原子級的分辨率預測。
將語言模型的參數擴大到15B,就可以系統性地研究規模對蛋白質結構學習的影響。
我們看到,蛋白質結構預測的非線性曲線是模型規模的函數,並且觀察到了語言模型對序列的理解程度與結構預測之間的強烈聯繫。
ESM-2系列的模型是迄今為止訓練的最大的蛋白質語言模型,其參數僅比最近開發的最大文字模型少一個數量級。
而且,ESM-2比以前的模型有非常大的改進,即使在150M的參數下,ESM-2也比ESM-1代語言模型在6.5億的參數下捕捉到更準確的結構圖。
研究人員表示,ESMFold效能的最大驅動力是語言模型。由於語言模型的迷惑性和結構預測的準確性之間有很強的聯繫,他們發現當ESM-2能較好地理解蛋白質序列時,就可以獲得與目前最先進的模型相當的預測結果。
ESMFold獲得了準確的原子解析度結構預測,推理時間也比AlphaFold2快了一個數量級。
在實踐中,速度的優勢甚至還要更大。因為ESMFold不需要搜尋和演化相關的序列來建構MSA。
雖然說有更快的方法可以減少搜尋時間,但再怎麼減少還是可能會很長。
而推理時間的極大縮短帶來的利好不言自明——速度的提高將使繪製大型元基因組學序列資料庫的結構空間成為可能。
除了結構為基礎的工具來辨識遠端同源性和保護性之外,用ESMFold進行快速且準確的結構預測,還能在大量新序列集合的結構和功能分析中發揮重要作用。
在有限的時間內獲得數以百萬計的預測結構,有利於發現對天然蛋白質的廣度和多樣性的新認識,並能發現全新的蛋白質結構和蛋白質功能。
作者介紹
本文的共同一作是來自Meta AI的Zeming Lin。

根據個人主頁介紹,Zeming在紐約大學攻讀博士學位,並在Meta AI擔任研究工程師(訪問),主要負責後端基礎設施的工作。
他本碩都就讀於維吉尼亞大學,在那裡,他和Yanjun Qi大佬一起做有關機器學習應用的研究,尤其是在蛋白質結構預測方面。
感興趣的領域為深度學習、結構預測,以及資訊生物學。
以上是快了一個0! Meta祭出150億參數蛋白質大模型,碾壓AlphaFold2的詳細內容。更多資訊請關注PHP中文網其他相關文章!

熱AI工具

Undresser.AI Undress
人工智慧驅動的應用程序,用於創建逼真的裸體照片

AI Clothes Remover
用於從照片中去除衣服的線上人工智慧工具。

Undress AI Tool
免費脫衣圖片

Clothoff.io
AI脫衣器

Video Face Swap
使用我們完全免費的人工智慧換臉工具,輕鬆在任何影片中換臉!

熱門文章
熱工具

記事本++7.3.1
好用且免費的程式碼編輯器

SublimeText3漢化版
中文版,非常好用

禪工作室 13.0.1
強大的PHP整合開發環境

Dreamweaver CS6
視覺化網頁開發工具

SublimeText3 Mac版
神級程式碼編輯軟體(SublimeText3)



 DeepMind機器人打乒乓球,正手、反手溜到飛起,全勝人類初學者
Aug 09, 2024 pm 04:01 PM
DeepMind機器人打乒乓球,正手、反手溜到飛起,全勝人類初學者
Aug 09, 2024 pm 04:01 PM
但可能打不過公園裡的老大爺?巴黎奧運正在如火如荼地進行中,乒乓球項目備受關注。同時,機器人打乒乓球也取得了新突破。剛剛,DeepMind提出了第一個在競技乒乓球比賽中達到人類業餘選手等級的學習型機器人智能體。論文地址:https://arxiv.org/pdf/2408.03906DeepMind這個機器人打乒乓球什麼程度呢?大概和人類業餘選手不相上下:正手反手都會:對手採用多種打法,機器人也能招架得住:接不同旋轉的發球:不過,比賽激烈程度似乎不如公園老大爺對戰。對機器人來說,乒乓球運動
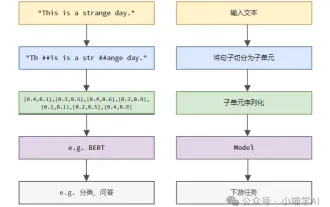 一文搞懂Tokenization!
Apr 12, 2024 pm 02:31 PM
一文搞懂Tokenization!
Apr 12, 2024 pm 02:31 PM
語言模型是對文字進行推理的,文字通常是字串形式,但模型的輸入只能是數字,因此需要將文字轉換成數字形式。 Tokenization是自然語言處理的基本任務,根據特定需求能夠把一段連續的文字序列(如句子、段落等)切分為一個字元序列(如單字、片語、字元、標點等多個單元),其中的單元稱為token或詞語。根據下圖所示的具體流程,首先將文字句子切分成一個個單元,然後將單元素數值化(映射為向量),再將這些向量輸入到模型進行編碼,最後輸出到下游任務進一步得到最終的結果。文本切分依照文本切分的粒度可以將Toke
 雲端部署大模型的三個秘密
Apr 24, 2024 pm 03:00 PM
雲端部署大模型的三個秘密
Apr 24, 2024 pm 03:00 PM
編譯|星璇出品|51CTO技術堆疊(微訊號:blog51cto)在過去的兩年裡,我更參與了使用大型語言模型(LLMs)的生成AI項目,而非傳統的系統。我開始懷念無伺服器雲端運算。它們的應用範圍廣泛,從增強對話AI到為各行各業提供複雜的分析解決方案,以及其他許多功能。許多企業將這些模型部署在雲端平台上,因為公有雲供應商已經提供了現成的生態系統,而且這是阻力最小的路徑。然而,這並不便宜。雲端還提供了其他好處,如可擴展性、效率和高階運算能力(按需提供GPU)。在公有雲平台上部署LLM的過程有一些鮮為人知的
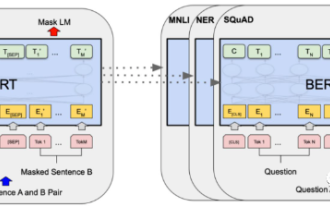 大規模語言模型高效參數微調--BitFit/Prefix/Prompt 微調系列
Oct 07, 2023 pm 12:13 PM
大規模語言模型高效參數微調--BitFit/Prefix/Prompt 微調系列
Oct 07, 2023 pm 12:13 PM
2018年Google發布了BERT,一經面世便一舉擊敗11個NLP任務的State-of-the-art(Sota)結果,成為了NLP界新的里程碑;BERT的結構如下圖所示,左邊是BERT模型預訓練過程,右邊是對於具體任務的微調過程。其中,微調階段是後續用於一些下游任務的時候進行微調,例如:文本分類,詞性標註,問答系統等,BERT無需調整結構就可以在不同的任務上進行微調。透過」預訓練語言模型+下游任務微調」的任務設計,帶來了強大的模型效果。從此,「預訓練語言模型+下游任務微調」便成為了NLP領域主流訓
 AlphaFold 3 重磅問世,全面預測蛋白質與所有生命分子相互作用及結構,準確度遠超以往水平
Jul 16, 2024 am 12:08 AM
AlphaFold 3 重磅問世,全面預測蛋白質與所有生命分子相互作用及結構,準確度遠超以往水平
Jul 16, 2024 am 12:08 AM
编辑|萝卜皮自2021年发布强大的AlphaFold2以来,科学家们一直在使用蛋白质结构预测模型来绘制细胞内各种蛋白质结构的图谱、发现药物,并绘制每种已知蛋白质相互作用的「宇宙图」。就在刚刚,GoogleDeepMind发布了AlphaFold3模型,该模型能够对包括蛋白质、核酸、小分子、离子和修饰残基在内的复合物进行联合结构预测。AlphaFold3的准确性对比过去许多专用工具(蛋白质-配体相互作用、蛋白质-核酸相互作用、抗体-抗原预测)有显著提高。这表明,在单个统一的深度学习框架内,可以实现
 為大模型提供全新科學複雜問答基準與評估體系,UNSW、阿貢、芝加哥大學等多家機構共同推出SciQAG框架
Jul 25, 2024 am 06:42 AM
為大模型提供全新科學複雜問答基準與評估體系,UNSW、阿貢、芝加哥大學等多家機構共同推出SciQAG框架
Jul 25, 2024 am 06:42 AM
編輯|ScienceAI問答(QA)資料集在推動自然語言處理(NLP)研究中發揮著至關重要的作用。高品質QA資料集不僅可以用於微調模型,也可以有效評估大語言模型(LLM)的能力,尤其是針對科學知識的理解和推理能力。儘管目前已有許多科學QA數據集,涵蓋了醫學、化學、生物等領域,但這些數據集仍有一些不足之處。其一,資料形式較為單一,大多數為多項選擇題(multiple-choicequestions),它們易於進行評估,但限制了模型的答案選擇範圍,無法充分測試模型的科學問題解答能力。相比之下,開放式問答
 Meta 推出 AI 語言模型 LLaMA,一個有著 650 億參數的大型語言模型
Apr 14, 2023 pm 06:58 PM
Meta 推出 AI 語言模型 LLaMA,一個有著 650 億參數的大型語言模型
Apr 14, 2023 pm 06:58 PM
2月25日消息,Meta在當地時間週五宣布,它將推出一種針對研究社區的基於人工智慧(AI)的新型大型語言模型,與微軟、谷歌等一眾受到ChatGPT刺激的公司一同加入人工智能競賽。 Meta的LLaMA是「大型語言模式MetaAI」(LargeLanguageModelMetaAI)的縮寫,它可以在非商業許可下提供給政府、社區和學術界的研究人員和實體工作者。該公司將提供底層程式碼供用戶使用,因此用戶可以自行調整模型,並將其用於與研究相關的用例。 Meta表示,該模型對算力的要
 從頭開始構建,DeepMind新論文用偽代碼詳解Transformer
Apr 09, 2023 pm 08:31 PM
從頭開始構建,DeepMind新論文用偽代碼詳解Transformer
Apr 09, 2023 pm 08:31 PM
2017 年 Transformer 橫空出世,由Google在論文《Attention is all you need》中引入。這篇論文拋棄了以往深度學習任務裡面所使用的 CNN 和 RNN。這項開創性的研究顛覆了以往序列建模和 RNN 劃等號的思路,如今被廣泛用於 NLP。大熱的 GPT、BERT 等都是基於 Transformer 建構的。 Transformer 自推出以來,研究者已經提出了許多變體。但大家對 Transformer 的描述似乎都是以口頭形式、圖形解釋等方式介紹架構。關於 Tra
