

從'最罕見變異”中找到'最容易患病個體”,AI精準預測基因變異致病性

通常而言,幾乎每個人都會攜帶有幾十個潛在有害的罕見變異。無法有效識別可能患有疾病的高風險個體,是透過常見變異進行臨床評估所遇到的最大困難之一。在靈長類基因組計畫的研究中,科學家利用人工智慧神經網路PrimateAI-3D,以「用最罕見的變異找到最容易生病的個體」的思路,透過演化分析定位高致病性的罕見突變,並將其用於預測個體患病風險。
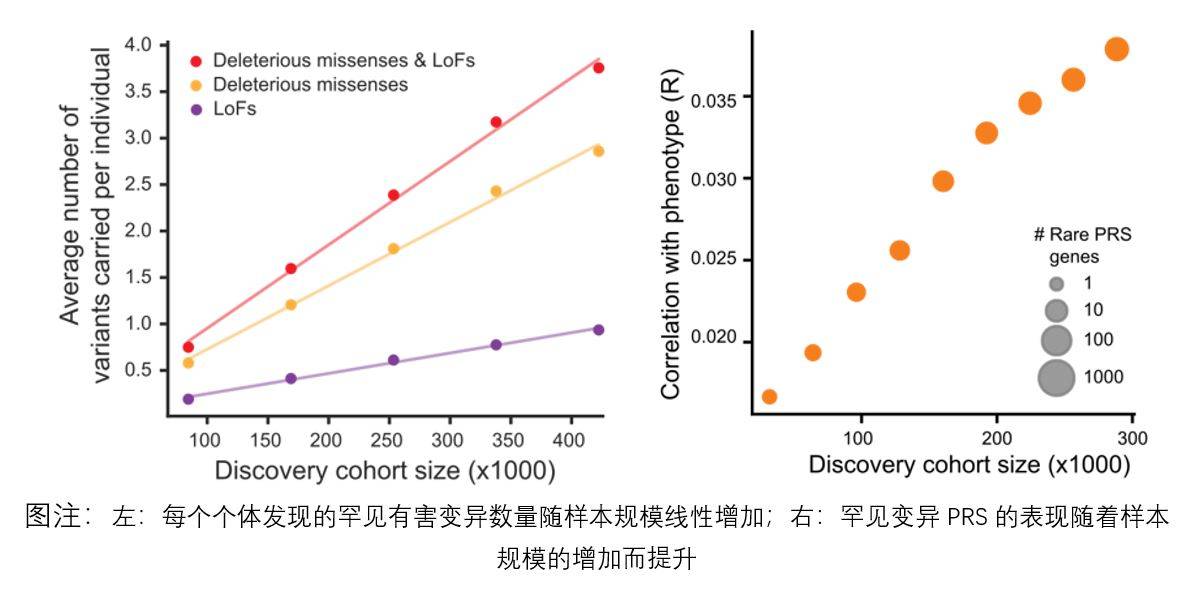
西班牙托馬斯·馬奎斯-博內特教授團隊、I l l umina人工智慧實驗室聯合多個課題組,透過對233種靈長類物種的共809個樣本進行全基因組測序數據比較,鑑定出人類直系同源蛋白上430萬個可能導致蛋白質結構變化的基因變異位點。
研究人員以上述基因變異位點為資料集基礎,將其加入人類疾病基因資料中,以包含450萬種可能造成良性變異的基因資料集訓練PrimateAI-3D人工智慧神經網絡,使其能夠更準確預測基因變異的致病性。
基因變異是導致疾病的最主要原因之一。基於非人靈長類與人類的親緣關係,相同的基因突變可能帶來相似的結果,在靈長類中常見的突變可能意味著這些變異更可能是無害或危害極其有限的。
那麼,如何預測一個人的遺傳因素對諸如糖尿病、心血管疾病等常見疾病帶來的患病風險呢?是用數千種影響較小的常見遺傳變異的總和來進行評估,還是用少數影響顯著的罕見突變的總和進行評估更好?
綜合研究表明,常見變異和罕見變異在預測人類疾病風險方面具有互補作用。常見的突變可以識別出更多可能患病的個體,而罕見的突變更容易發現最高風險的異常個體。因此,在臨床評估上納入罕見變異可能比僅使用常見變異更能識別出極端個體,而這些極端個體才是大部分疾病的最終患者群體,也是最需要治療或遭受嚴重早期病變的群體,這對預防性篩檢具有重要意義。
這項研究成功展示了將靈長類群體定序資料與深度學習模式結合的應用,有助於我們了解人類基因變異的致病性,可幫助個人化基因組醫學在臨床上提供更佳診斷指導。
作者:浙江大學張國捷教授主題組集體創作、整理
編輯:許琦敏
*文匯獨家稿件,轉載請註明出處。
以上是從'最罕見變異”中找到'最容易患病個體”,AI精準預測基因變異致病性的詳細內容。更多資訊請關注PHP中文網其他相關文章!

熱AI工具

Undresser.AI Undress
人工智慧驅動的應用程序,用於創建逼真的裸體照片

AI Clothes Remover
用於從照片中去除衣服的線上人工智慧工具。

Undress AI Tool
免費脫衣圖片

Clothoff.io
AI脫衣器

Video Face Swap
使用我們完全免費的人工智慧換臉工具,輕鬆在任何影片中換臉!

熱門文章
熱工具

記事本++7.3.1
好用且免費的程式碼編輯器

SublimeText3漢化版
中文版,非常好用

禪工作室 13.0.1
強大的PHP整合開發環境

Dreamweaver CS6
視覺化網頁開發工具

SublimeText3 Mac版
神級程式碼編輯軟體(SublimeText3)



 10個生成AI編碼擴展,在VS代碼中,您必須探索
Apr 13, 2025 am 01:14 AM
10個生成AI編碼擴展,在VS代碼中,您必須探索
Apr 13, 2025 am 01:14 AM
嘿,編碼忍者!您當天計劃哪些與編碼有關的任務?在您進一步研究此博客之前,我希望您考慮所有與編碼相關的困境,這是將其列出的。 完畢? - 讓&#8217
 GPT-4O vs OpenAI O1:新的Openai模型值得炒作嗎?
Apr 13, 2025 am 10:18 AM
GPT-4O vs OpenAI O1:新的Openai模型值得炒作嗎?
Apr 13, 2025 am 10:18 AM
介紹 Openai已根據備受期待的“草莓”建築發布了其新模型。這種稱為O1的創新模型增強了推理能力,使其可以通過問題進行思考
 視覺語言模型(VLMS)的綜合指南
Apr 12, 2025 am 11:58 AM
視覺語言模型(VLMS)的綜合指南
Apr 12, 2025 am 11:58 AM
介紹 想像一下,穿過美術館,周圍是生動的繪畫和雕塑。現在,如果您可以向每一部分提出一個問題並獲得有意義的答案,該怎麼辦?您可能會問:“您在講什麼故事?
 pixtral -12b:Mistral AI'第一個多模型模型 - 分析Vidhya
Apr 13, 2025 am 11:20 AM
pixtral -12b:Mistral AI'第一個多模型模型 - 分析Vidhya
Apr 13, 2025 am 11:20 AM
介紹 Mistral發布了其第一個多模式模型,即Pixtral-12b-2409。該模型建立在Mistral的120億參數Nemo 12B之上。是什麼設置了該模型?現在可以拍攝圖像和Tex
 如何在SQL中添加列? - 分析Vidhya
Apr 17, 2025 am 11:43 AM
如何在SQL中添加列? - 分析Vidhya
Apr 17, 2025 am 11:43 AM
SQL的Alter表語句:動態地將列添加到數據庫 在數據管理中,SQL的適應性至關重要。 需要即時調整數據庫結構嗎? Alter表語句是您的解決方案。本指南的詳細信息添加了Colu
 超越駱駝戲:大型語言模型的4個新基準
Apr 14, 2025 am 11:09 AM
超越駱駝戲:大型語言模型的4個新基準
Apr 14, 2025 am 11:09 AM
陷入困境的基準:駱駝案例研究 2025年4月上旬,梅塔(Meta)揭開了其Llama 4套件的模特,擁有令人印象深刻的性能指標,使他們對GPT-4O和Claude 3.5 Sonnet等競爭對手的良好定位。倫斯的中心
 如何使用AGNO框架構建多模式AI代理?
Apr 23, 2025 am 11:30 AM
如何使用AGNO框架構建多模式AI代理?
Apr 23, 2025 am 11:30 AM
在從事代理AI時,開發人員經常發現自己在速度,靈活性和資源效率之間進行權衡。我一直在探索代理AI框架,並遇到了Agno(以前是Phi-
 多動症遊戲,健康工具和AI聊天機器人如何改變全球健康
Apr 14, 2025 am 11:27 AM
多動症遊戲,健康工具和AI聊天機器人如何改變全球健康
Apr 14, 2025 am 11:27 AM
視頻遊戲可以緩解焦慮,建立焦點或支持多動症的孩子嗎? 隨著醫療保健在全球範圍內挑戰,尤其是在青年中的挑戰,創新者正在轉向一種不太可能的工具:視頻遊戲。現在是世界上最大的娛樂印度河之一
