

AI未經學習!最新研究揭示了解讀人工智慧黑盒的方法

人工智慧(AI)一直在迅速發展,但對人類來說,強大的模型卻是個「黑盒子」。
我們不了解模型內部的運作原理,不清楚它得出結論的過程。
然而最近,波昂大學(University of Bonn)的化學資訊學專家Jürgen Bajorath教授和他的團隊取得了重大突破。
他們設計了一種技術,揭示了藥物研究中使用的某些人工智慧系統的運作機制。
研究顯示,人工智慧模型主要透過回憶現有數據來預測藥物有效性,而非學習特定化學交互作用。
-也就是說,AI預測純靠拼湊記憶,機器學習其實並沒有學習!
他們的研究結果最近發表在《自然機器智能》(Nature Machine Intelligence)雜誌上。

論文網址:https://www.nature.com/articles/s42256-023-00756-9
在醫藥領域,研究人員正在狂熱地尋找有效的活性物質來對抗疾病-哪種藥物分子最有效?
通常,這些有效的分子(化合物)會對接在蛋白質上,蛋白質作為觸發特定生理作用鏈的酵素或受體。
在特殊情況下,某些分子也負責阻斷體內的不良反應,例如過度的發炎反應。
可能的化合物數量龐大,尋找有效的化合物就像大海撈針一樣。
因此,研究人員首先使用AI模型來預測,哪些分子最能與各自的目標蛋白對接並牢固結合。然後在實驗研究中,更詳細地進一步篩選這些候選藥物。
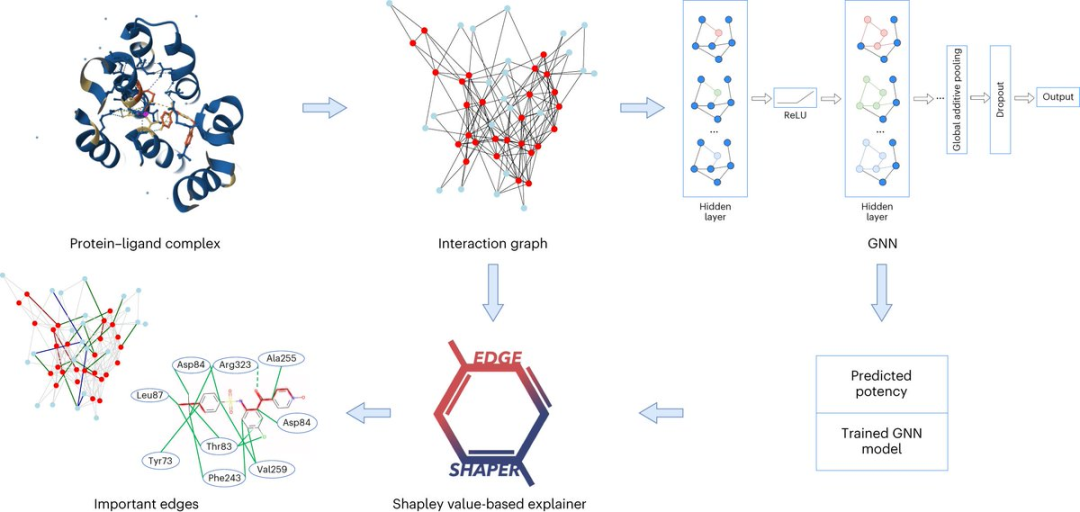
自人工智慧發展以來,藥物發現研究也越來越多地採用AI相關的技術。
比如圖神經網路(GNN),適用於預測某種分子與目標蛋白結合的強度。
圖由表示物件的節點和表示節點之間關係的邊組成。在蛋白質與配體複合物的圖表示中,圖的邊連接蛋白質或配體節點,表示物質的結構,或蛋白質和配體之間的相互作用。
GNN模型使用從X射線結構中提取的蛋白質配體相互作用圖,來預測配體親和力。
Jürgen Bajorath教授表示,GNN模型對我們來說就像一個黑盒子,我們無法得知它如何得出自己的預測。

Jürgen Bajorath教授任職於波昂大學LIMES研究所、波昂-亞琛國際資訊科技中心(Bonn-Aachen International Center for Information Technology)和拉瑪機器學習與人工智慧研究所(Lamarr Institute for Machine Learning and Artificial Intelligence)。
人工智慧如何運作?
來自波昂大學化學資訊學的研究人員,與羅馬Sapienza大學的同事一起,詳細分析了圖神經網路是否真的學習到了蛋白質與配體的相互作用。
研究人員使用他們專門開發的「EdgeSHAPer」方法分析了總共六種不同的GNN架構。
EdgeSHAPer程式可以判斷GNN是否學習了化合物和蛋白質之間最重要的相互作用,或者是透過其他的方式來得出預測。
科學家們使用從蛋白質配體複合物結構中提取的圖訓練了六個GNN,——化合物的作用方式以及與目標蛋白的結合強度已知。
然後,在其他複合物上測試經過訓練的GNN,並使用EdgeSHAPer分析GNN如何產生預測。
「如果GNN按照預期行事,它們需要學習化合物和目標蛋白之間的相互作用,並且透過優先考慮特定的相互作用來給出預測」。
然而,根據研究小組的分析,六個GNN基本上都沒有做到這一點。大多數GNN只學會了一些蛋白質與藥物的相互作用,主要集中在配體上。
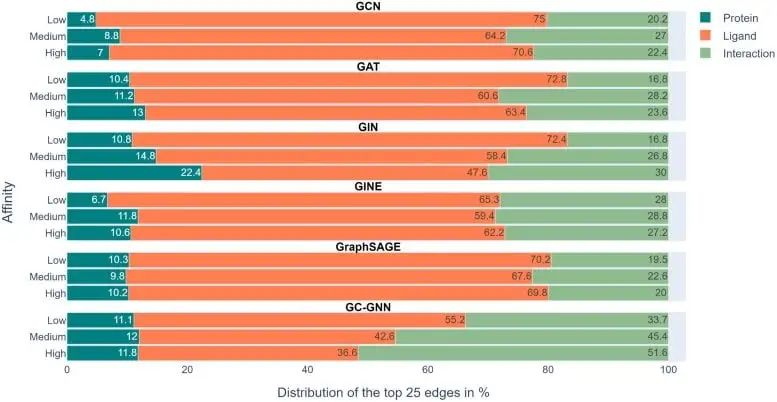
上圖展示了6個GNN中的實驗結果,色標條表示用EdgeSHAPer確定的每個預測的前25個邊中蛋白質、配體和相互作用所佔的平均比例。
我們可以看到,代表綠色的相互作用本該是模型需要學到的,然而在整個實驗中所佔的比例都不高,而代表配體的橙色條佔了最大的比例。
為了預測分子與目標蛋白的結合強度,模型主要「記住」了它們在訓練過程中遇到的化學相似分子及其結合數據,而不管目標蛋白如何。這些被記住的化學相似性基本上決定了預測。
這讓人想起「聰明的漢斯效應」(Clever Hans effect),--就像那匹看起來會數數的馬,實際上是根據同伴面部表情和手勢的細微差別,來推斷出預期的結果。
這或許意味著,GNN所謂的「學習能力」可能是站不住腳的,模型的預測在很大程度上被高估了,因為可以使用化學知識和更簡單的方法進行同等品質的預測。
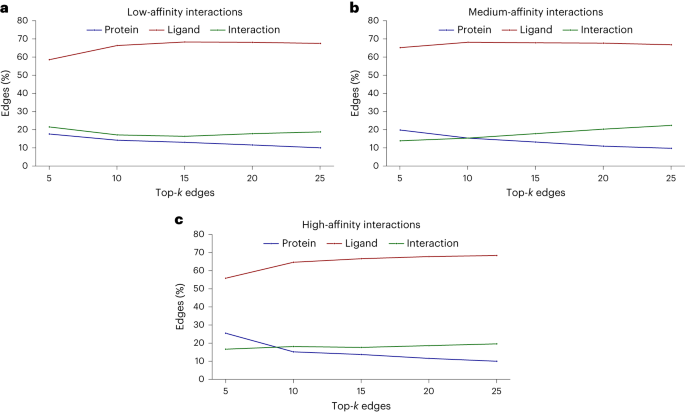
不過,研究中也發現了另一個現象:當測試化合物的效力增加時,模型傾向於學習到更多的相互作用。
也許透過修改表徵和訓練技術,這些GNN還能朝著理想的方向進一步改進。不過,對於可以根據分子圖學習物理量的假設,一般來說應該要謹慎看待。
「人工智慧不是黑魔法。」
#以上是AI未經學習!最新研究揭示了解讀人工智慧黑盒的方法的詳細內容。更多資訊請關注PHP中文網其他相關文章!

熱AI工具

Undresser.AI Undress
人工智慧驅動的應用程序,用於創建逼真的裸體照片

AI Clothes Remover
用於從照片中去除衣服的線上人工智慧工具。

Undress AI Tool
免費脫衣圖片

Clothoff.io
AI脫衣器

Video Face Swap
使用我們完全免費的人工智慧換臉工具,輕鬆在任何影片中換臉!

熱門文章
熱工具

記事本++7.3.1
好用且免費的程式碼編輯器

SublimeText3漢化版
中文版,非常好用

禪工作室 13.0.1
強大的PHP整合開發環境

Dreamweaver CS6
視覺化網頁開發工具

SublimeText3 Mac版
神級程式碼編輯軟體(SublimeText3)



 如何理解C 中的DMA操作?
Apr 28, 2025 pm 10:09 PM
如何理解C 中的DMA操作?
Apr 28, 2025 pm 10:09 PM
DMA在C 中是指DirectMemoryAccess,直接內存訪問技術,允許硬件設備直接與內存進行數據傳輸,不需要CPU干預。 1)DMA操作高度依賴於硬件設備和驅動程序,實現方式因係統而異。 2)直接訪問內存可能帶來安全風險,需確保代碼的正確性和安全性。 3)DMA可提高性能,但使用不當可能導致系統性能下降。通過實踐和學習,可以掌握DMA的使用技巧,在高速數據傳輸和實時信號處理等場景中發揮其最大效能。
 C 中的chrono庫如何使用?
Apr 28, 2025 pm 10:18 PM
C 中的chrono庫如何使用?
Apr 28, 2025 pm 10:18 PM
使用C 中的chrono庫可以讓你更加精確地控制時間和時間間隔,讓我們來探討一下這個庫的魅力所在吧。 C 的chrono庫是標準庫的一部分,它提供了一種現代化的方式來處理時間和時間間隔。對於那些曾經飽受time.h和ctime折磨的程序員來說,chrono無疑是一個福音。它不僅提高了代碼的可讀性和可維護性,還提供了更高的精度和靈活性。讓我們從基礎開始,chrono庫主要包括以下幾個關鍵組件:std::chrono::system_clock:表示系統時鐘,用於獲取當前時間。 std::chron
 怎樣在C 中測量線程性能?
Apr 28, 2025 pm 10:21 PM
怎樣在C 中測量線程性能?
Apr 28, 2025 pm 10:21 PM
在C 中測量線程性能可以使用標準庫中的計時工具、性能分析工具和自定義計時器。 1.使用庫測量執行時間。 2.使用gprof進行性能分析,步驟包括編譯時添加-pg選項、運行程序生成gmon.out文件、生成性能報告。 3.使用Valgrind的Callgrind模塊進行更詳細的分析,步驟包括運行程序生成callgrind.out文件、使用kcachegrind查看結果。 4.自定義計時器可靈活測量特定代碼段的執行時間。這些方法幫助全面了解線程性能,並優化代碼。
 量化交易所排行榜2025 數字貨幣量化交易APP前十名推薦
Apr 30, 2025 pm 07:24 PM
量化交易所排行榜2025 數字貨幣量化交易APP前十名推薦
Apr 30, 2025 pm 07:24 PM
交易所內置量化工具包括:1. Binance(幣安):提供Binance Futures量化模塊,低手續費,支持AI輔助交易。 2. OKX(歐易):支持多賬戶管理和智能訂單路由,提供機構級風控。獨立量化策略平台有:3. 3Commas:拖拽式策略生成器,適用於多平台對沖套利。 4. Quadency:專業級算法策略庫,支持自定義風險閾值。 5. Pionex:內置16 預設策略,低交易手續費。垂直領域工具包括:6. Cryptohopper:雲端量化平台,支持150 技術指標。 7. Bitsgap:
 怎樣在C 中處理高DPI顯示?
Apr 28, 2025 pm 09:57 PM
怎樣在C 中處理高DPI顯示?
Apr 28, 2025 pm 09:57 PM
在C 中處理高DPI顯示可以通過以下步驟實現:1)理解DPI和縮放,使用操作系統API獲取DPI信息並調整圖形輸出;2)處理跨平台兼容性,使用如SDL或Qt的跨平台圖形庫;3)進行性能優化,通過緩存、硬件加速和動態調整細節級別來提升性能;4)解決常見問題,如模糊文本和界面元素過小,通過正確應用DPI縮放來解決。
 C 中的實時操作系統編程是什麼?
Apr 28, 2025 pm 10:15 PM
C 中的實時操作系統編程是什麼?
Apr 28, 2025 pm 10:15 PM
C 在實時操作系統(RTOS)編程中表現出色,提供了高效的執行效率和精確的時間管理。 1)C 通過直接操作硬件資源和高效的內存管理滿足RTOS的需求。 2)利用面向對象特性,C 可以設計靈活的任務調度系統。 3)C 支持高效的中斷處理,但需避免動態內存分配和異常處理以保證實時性。 4)模板編程和內聯函數有助於性能優化。 5)實際應用中,C 可用於實現高效的日誌系統。
 給MySQL表添加和刪除字段的操作步驟
Apr 29, 2025 pm 04:15 PM
給MySQL表添加和刪除字段的操作步驟
Apr 29, 2025 pm 04:15 PM
在MySQL中,添加字段使用ALTERTABLEtable_nameADDCOLUMNnew_columnVARCHAR(255)AFTERexisting_column,刪除字段使用ALTERTABLEtable_nameDROPCOLUMNcolumn_to_drop。添加字段時,需指定位置以優化查詢性能和數據結構;刪除字段前需確認操作不可逆;使用在線DDL、備份數據、測試環境和低負載時間段修改表結構是性能優化和最佳實踐。
 C 中的字符串流如何使用?
Apr 28, 2025 pm 09:12 PM
C 中的字符串流如何使用?
Apr 28, 2025 pm 09:12 PM
C 中使用字符串流的主要步驟和注意事項如下:1.創建輸出字符串流並轉換數據,如將整數轉換為字符串。 2.應用於復雜數據結構的序列化,如將vector轉換為字符串。 3.注意性能問題,避免在處理大量數據時頻繁使用字符串流,可考慮使用std::string的append方法。 4.注意內存管理,避免頻繁創建和銷毀字符串流對象,可以重用或使用std::stringstream。
