

快了一个0!Meta祭出150亿参数蛋白质大模型,碾压AlphaFold2

迄今为止规模最大的蛋白质语言模型问世了!
一年前,DeepMind开源AlphaFold2连登Nature、Science,刷爆生物和AI学界。
一年后,Meta带着速度快一个数量级的ESMFold来了。
不光速度快,模型还足足有150亿个参数。

LeCun发推称赞,这是Meta-FAIR蛋白质团队的伟大新成果。

共同一作Zeming Lin透露,30亿参数的大模型在256个GPU上训练了3个星期,而ESMfold在128个GPU上用了10天。至于150亿参数的版本,目前还不清楚。
他还表示,代码随后肯定会开源,敬请关注!
又大又快!
今天,我们的主角是ESMFold,一个从蛋白质个体的序列,直接进行高准确度、端对端、原子层级结构预测的模型。

论文地址:https://www.biorxiv.org/content/10.1101/2022.07.20.500902v1
150亿个参数带来的好处不必多说——通过训练,如今的大模型可以在原子大小的精度上预测蛋白质的三维结构。
从准确度上看,ESMFold和AlphaFold2、RoseTTAFold差不多。
但是,ESMFold推测速度要比AlphaFold2快一个数量级!
一下说数量级可能不好理解三者之间速度的对比,看看下面这张图就懂了。

有啥区别?
虽说AlphaFold2和RoseTTAFold在原子分辨率结构预测问题上取得了突破性的成功,但它们也依赖于使用多序列比对(MSA)和类似的蛋白质结构模板来实现最佳性能。
相比之下,通过利用语言模型的内部表征,ESMFold只用一个序列作为输入就能生成相应的结构预测,从而大大加快了结构预测的速度。

研究人员发现,ESMFold对低复杂度序列的预测与当下最先进的模型相当。
而且,结构预测的准确性与语言模型的复杂度息息相关,也就是说,当语言模型能更好地理解序列时,便可以更好地理解结构。

目前,有数十亿结构和功能未知的蛋白质序列,其中许多来自元基因组测序。
利用ESMFold,研究人员只需6个小时,就能折叠完成100万个元基因组序列的随机样本。

其中很大一部分具有高置信度,并且与任何已知的结构不同(在数据库中没有记录)。
研究人员认为,ESMFold可以帮助理解那些超出现有认知的蛋白质结构。

此外,由于ESMFold的预测速度比现有的模型快一个数量级,因此研究人员便可借助ESMFold来协助填补快速增长的蛋白质序列数据库与进展缓慢的蛋白质结构和功能数据库之间的鸿沟。
150亿参数的蛋白质语言模型
接下来我们就来具体说说Meta这款全新的ESMFold。
ESM-2是一个基于Transformer的语言模型,并使用注意力机制来学习输入序列中成对氨基酸之间的相互作用模式。
相对于上一代模型ESM-1b,Meta对模型结构、训练参数进行了改进,并增加了计算资源和数据。同时,相对位置嵌入的加入,使模型能够推广到任意长度的序列。
从结果来看,具有1.5亿个参数的ESM-2模型比具有6.5亿个参数的ESM-1b模型表现得更好。
此外,在结构预测的基准上,ESM-2也超过了其他的蛋白质语言模型。这种性能的提高与大型语言建模领域建立的规律是一致的。

随着ESM-2规模的增加,可以观察到语言建模的精度有很大的提高。

端到端的单序列结构预测
SMFold和AlphaFold2的一个关键区别是,ESMFold使用语言模型表示,消除了对明确的同源序列(以MSA的形式)作为输入的需要。
ESMFold通过用一个处理序列的Transformer模块取代处理MSA的计算昂贵的网络模块,简化了AlphaFold2中的Evoformer。这种简化意味着ESMFold的速度大大提高,远高于基于MSA的模型。
折叠主干的输出接下来又被一个结构模块处理,它负责输出最终的原子级结构和预测的置信度。

研究人员将ESMFold与AlphaFold2和RoseTTAFold在CAMEO(2022年4月至2022年6月)和CASP14(2020年5月)测试集上进行比较。
当只给单一序列输入时,ESMFold的表现要比Alphafold 2好得多。
而当使用完整的管道时,AlphaFold2在CAMEO和CASP14上分别达到了88.3和84.7。ESMFold在CAMEO上取得了与RoseTTAfold相当的准确率,其平均TM分数为82.0。

结论
研究人员发现,以无监督学习为目标的语言模型在一个大型的进化多样化的蛋白质序列数据库中训练,能够对蛋白质结构进行原子级的分辨率预测。
将语言模型的参数扩大到15B,就可以系统地研究规模对蛋白质结构学习的影响。
我们看到,蛋白质结构预测的非线性曲线是模型规模的函数,并且观察到了语言模型对序列的理解程度与结构预测之间的强烈联系。
ESM-2系列的模型是迄今为止训练的最大的蛋白质语言模型,其参数仅比最近开发的最大文本模型少一个数量级。
而且,ESM-2比以前的模型有非常大的改进,即使在150M的参数下,ESM-2也比ESM-1代语言模型在6.5亿的参数下捕捉到更准确的结构图。
研究人员表示,ESMFold性能的最大驱动力是语言模型。由于语言模型的迷惑性和结构预测的准确性之间有很强的联系,他们发现当ESM-2能较好地理解蛋白质序列时,就可以获得与目前最先进的模型相当的预测结果。
ESMFold获得了准确的原子分辨率结构预测,推理时间还比AlphaFold2快了一个数量级。
在实践中,速度的优势甚至还要更大。因为ESMFold不需要搜索和进化相关的序列来构建MSA。
虽说有更快的方法可以减少搜索时间,但再怎么减少还是可能会很长。
而推理时间的极大缩短带来的利好不言自明——速度的提高将使绘制大型元基因组学序列数据库的结构空间成为可能。
除了基于结构的工具来识别远端同源性和保护性之外,用ESMFold进行快速准确的结构预测,还能在大量新序列集合的结构和功能分析中发挥重要作用。
在有限的时间内获得数以百万计的预测结构,有利于发现对天然蛋白质的广度和多样性的新认识,并能发现全新的蛋白质结构和蛋白质功能。
作者介绍
本文的共同一作是来自Meta AI的Zeming Lin。

据个人主页介绍,Zeming在纽约大学攻读博士学位,并在Meta AI担任研究工程师(访问),主要负责后端基础设施的工作。
他本硕都就读于弗吉尼亚大学,在那里,他和Yanjun Qi大佬一同做有关机器学习应用的研究,尤其是在蛋白质结构预测方面。
感兴趣的领域为深度学习、结构预测,以及信息生物学。
以上是快了一个0!Meta祭出150亿参数蛋白质大模型,碾压AlphaFold2的详细内容。更多信息请关注PHP中文网其他相关文章!

热AI工具

Undresser.AI Undress
人工智能驱动的应用程序,用于创建逼真的裸体照片

AI Clothes Remover
用于从照片中去除衣服的在线人工智能工具。

Undress AI Tool
免费脱衣服图片

Clothoff.io
AI脱衣机

AI Hentai Generator
免费生成ai无尽的。

热门文章
热工具

记事本++7.3.1
好用且免费的代码编辑器

SublimeText3汉化版
中文版,非常好用

禅工作室 13.0.1
功能强大的PHP集成开发环境

Dreamweaver CS6
视觉化网页开发工具

SublimeText3 Mac版
神级代码编辑软件(SublimeText3)
热门话题



 DeepMind机器人打乒乓球,正手、反手溜到飞起,全胜人类初学者
Aug 09, 2024 pm 04:01 PM
DeepMind机器人打乒乓球,正手、反手溜到飞起,全胜人类初学者
Aug 09, 2024 pm 04:01 PM
但可能打不过公园里的老大爷?巴黎奥运会正在如火如荼地进行中,乒乓球项目备受关注。与此同时,机器人打乒乓球也取得了新突破。刚刚,DeepMind提出了第一个在竞技乒乓球比赛中达到人类业余选手水平的学习型机器人智能体。论文地址:https://arxiv.org/pdf/2408.03906DeepMind这个机器人打乒乓球什么水平呢?大概和人类业余选手不相上下:正手反手都会:对手采用多种打法,该机器人也能招架得住:接不同旋转的发球:不过,比赛激烈程度似乎不如公园老大爷对战。对机器人来说,乒乓球运动
 一文搞懂Tokenization!
Apr 12, 2024 pm 02:31 PM
一文搞懂Tokenization!
Apr 12, 2024 pm 02:31 PM
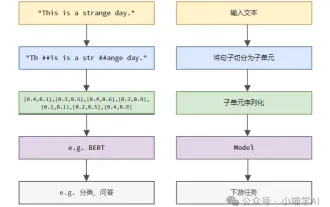
语言模型是对文本进行推理的,文本通常是字符串形式,但模型的输入只能是数字,因此需要将文本转换成数字形式。Tokenization是自然语言处理的基本任务,根据特定需求能够把一段连续的文本序列(如句子、段落等)切分为一个字符序列(如单词、短语、字符、标点等多个单元),其中的单元称为token或词语。根据下图所示的具体流程,首先将文本句子切分成一个个单元,然后将单元素数值化(映射为向量),再将这些向量输入到模型进行编码,最后输出到下游任务进一步得到最终的结果。文本切分按照文本切分的粒度可以将Toke
 大规模语言模型高效参数微调--BitFit/Prefix/Prompt 微调系列
Oct 07, 2023 pm 12:13 PM
大规模语言模型高效参数微调--BitFit/Prefix/Prompt 微调系列
Oct 07, 2023 pm 12:13 PM
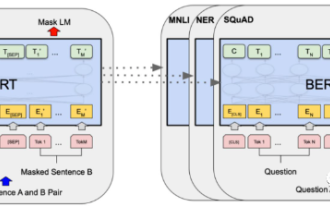
2018年谷歌发布了BERT,一经面世便一举击败11个NLP任务的State-of-the-art(Sota)结果,成为了NLP界新的里程碑;BERT的结构如下图所示,左边是BERT模型预训练过程,右边是对于具体任务的微调过程。其中,微调阶段是后续用于一些下游任务的时候进行微调,例如:文本分类,词性标注,问答系统等,BERT无需调整结构就可以在不同的任务上进行微调。通过”预训练语言模型+下游任务微调”的任务设计,带来了强大的模型效果。从此,“预训练语言模型+下游任务微调”便成为了NLP领域主流训
 为大模型提供全新科学复杂问答基准与测评体系,UNSW、阿贡、芝加哥大学等多家机构联合推出SciQAG框架
Jul 25, 2024 am 06:42 AM
为大模型提供全新科学复杂问答基准与测评体系,UNSW、阿贡、芝加哥大学等多家机构联合推出SciQAG框架
Jul 25, 2024 am 06:42 AM
编辑|ScienceAI问答(QA)数据集在推动自然语言处理(NLP)研究发挥着至关重要的作用。高质量QA数据集不仅可以用于微调模型,也可以有效评估大语言模型(LLM)的能力,尤其是针对科学知识的理解和推理能力。尽管当前已有许多科学QA数据集,涵盖了医学、化学、生物等领域,但这些数据集仍存在一些不足。其一,数据形式较为单一,大多数为多项选择题(multiple-choicequestions),它们易于进行评估,但限制了模型的答案选择范围,无法充分测试模型的科学问题解答能力。相比之下,开放式问答
 云端部署大模型的三个秘密
Apr 24, 2024 pm 03:00 PM
云端部署大模型的三个秘密
Apr 24, 2024 pm 03:00 PM
编译|星璇出品|51CTO技术栈(微信号:blog51cto)在过去的两年里,我更多地参与了使用大型语言模型(LLMs)的生成AI项目,而非传统的系统。我开始怀念无服务器云计算。它们的应用范围广泛,从增强对话AI到为各行各业提供复杂的分析解决方案,以及其他许多功能。许多企业将这些模型部署在云平台上,因为公共云提供商已经提供了现成的生态系统,而且这是阻力最小的路径。然而,这并不便宜。云还提供了其他好处,如可扩展性、效率和高级计算能力(按需提供GPU)。在公共云平台上部署LLM的过程有一些鲜为人知的
 AlphaFold 3 重磅问世,全面预测蛋白质与所有生命分子相互作用及结构,准确性远超以往水平
Jul 16, 2024 am 12:08 AM
AlphaFold 3 重磅问世,全面预测蛋白质与所有生命分子相互作用及结构,准确性远超以往水平
Jul 16, 2024 am 12:08 AM
编辑|萝卜皮自2021年发布强大的AlphaFold2以来,科学家们一直在使用蛋白质结构预测模型来绘制细胞内各种蛋白质结构的图谱、发现药物,并绘制每种已知蛋白质相互作用的「宇宙图」 。就在刚刚,GoogleDeepMind发布了AlphaFold3模型,该模型能够对包括蛋白质、核酸、小分子、离子和修饰残基在内的复合物进行联合结构预测。 AlphaFold3的准确性对比过去许多专用工具(蛋白质-配体相互作用、蛋白质-核酸相互作用、抗体-抗原预测)有显着提高。这表明,在单个统一的深度学习框架内,可以实现
 RoSA: 一种高效微调大模型参数的新方法
Jan 18, 2024 pm 05:27 PM
RoSA: 一种高效微调大模型参数的新方法
Jan 18, 2024 pm 05:27 PM
随着语言模型扩展到前所未有的规模,对下游任务进行全面微调变得十分昂贵。为了解决这个问题,研究人员开始关注并采用PEFT方法。PEFT方法的主要思想是将微调的范围限制在一小部分参数上,以降低计算成本,同时仍能实现自然语言理解任务的最先进性能。通过这种方式,研究人员能够在保持高性能的同时,节省计算资源,为自然语言处理领域带来新的研究热点。RoSA是一种新的PEFT技术,通过在一组基准测试的实验中,发现在使用相同参数预算的情况下,RoSA表现出优于先前的低秩自适应(LoRA)和纯稀疏微调方法。本文将深
 顺手训了一个史上超大ViT?Google升级视觉语言模型PaLI:支持100 种语言
Apr 12, 2023 am 09:31 AM
顺手训了一个史上超大ViT?Google升级视觉语言模型PaLI:支持100 种语言
Apr 12, 2023 am 09:31 AM
近几年自然语言处理的进展很大程度上都来自于大规模语言模型,每次发布的新模型都将参数量、训练数据量推向新高,同时也会对现有基准排行进行一次屠榜!比如今年4月,Google发布5400亿参数的语言模型PaLM(Pathways Language Model)在语言和推理类的一系列测评中成功超越人类,尤其是在few-shot小样本学习场景下的优异性能,也让PaLM被认为是下一代语言模型的发展方向。同理,视觉语言模型其实也是大力出奇迹,可以通过提升模型的规模来提升性能。当然了,如果只是多任务的视觉语言模
